Severe combined immunodeficiency (Severe combined immunodeficiency, SCID) is a class of diseases characterized by severe defects in both humoral and cellular immunity, generally with a more prominent T-cell immunodeficiency. It is a rare hereditary primary immunodeficiency disease.SCID is usually inherited in an autosomal recessive or X-linked manner, with some sporadic cases. About 50% of SCID patients have a positive family history. Most affected children present with long-term, recurrent, severe infections, including frequent pneumonia, chronic diarrhea, oral and skin candidiasis, otitis media, and growth and development disorders. A cure requires immunoreconstruction; if not treated promptly, most children will die within the first year of life.
SCID is characterized by congenital, hereditary abnormalities in B and T lymphocyte systems, which can be classified based on the quantity and function of T, B, and NK lymphocytes.
|
Disease |
Gene |
Lymphocyte Phenotype |
MOI |
||
|
T |
B |
NK |
|||
|
X-SCID |
IL2RG |
– |
+ |
– |
XLR |
|
JAK3-SCID (OMIM 600802) |
JAK3 |
AR |
|||
|
IL7R-SCID (OMIM 608971) |
IL7R |
– |
+ |
+ |
AR |
|
CD45 Deficiency (OMIM 608971) |
PTPRC (CD45) |
– |
+ |
+/– |
AR |
|
Adenosine Deaminase Deficiency |
ADA |
– |
– |
– |
AR |
|
RAG Deficiency Type SCID (OMIM 601457) |
RAG1 |
– |
– |
+ |
AR |
|
RAG2 |
– |
– |
+ |
||
|
SCID Alaskan (OMIM 602450) |
DCLRE1C |
– |
– |
+ |
AR |
|
TCR Deficiency |
CD3D, CD3E, CD247 |
–/Low |
+ |
+ |
AR |
|
DNAPKCS Deficiency (OMIM 615966) |
PRKDC |
– |
– |
+ |
AR |
|
Reticular Dysplasia (OMIM 267500) |
AK2 |
– |
– |
– |
AR |
|
CORO1a Deficiency (OMIM 615401) |
CORO1A |
–/Low |
+/– |
+/– |
AR |
This case mainly discusses the RAG deficiency SCID caused by mutations in the RAG1 and RAG2 genes. The recombination activating genes (RAGs) consist of RAG1 and RAG2 genes, and the proteins encoded by RAG1 and RAG2 are important DNA recombinases that work together to mediate the V(D)J rearrangement of lymphocyte antigen receptor genes, playing a key role in T and B cell differentiation and development, particularly in the formation of the antigen receptor diversity cell pool. Therefore, any obstruction in the expression of RAG1 or RAG2 leads to varying degrees of RAG immunodeficiency disease.
Pathogenesis

Based on the degree of impact of genetic abnormalities on RAG molecular activity, RAG deficiency disease can be divided into two main categories: RAG complete deficiency (rearrangement activity <1% of wild type) with no V(D)J rearrangement function and RAG residual deficiency (rearrangement activity >1% of wild type) with low activity V(D)J rearrangement function. RAG complete deficiency can be further classified into typical SCID and maternal T cell input type SCID based on immunophenotype and clinical characteristics. RAG residual deficiency can be divided into typical Omenn syndrome, atypical Omenn syndrome, granulomatous inflammation, γδ T cell dominant expansion type, and maternal T cell implantation.
In recent years, many types of immunodeficiency diseases have been reported, mostly due to viral infections. Although RAG deficiency disease can threaten the life of patients, proper diagnosis and early treatment measures such as bone marrow transplantation or umbilical cord blood stem cell transplantation can reduce mortality.
RAG is located on human chromosome 11p13 and consists of two adjacent genes, RAG1 and RAG2. The DNA recombinase RAG1 and RAG2 encoded by these two genes function as a complex, specifically recognizing and cutting the recombination signal sequences (RSS) located on both sides of the V(D)J gene segments of the antigen receptor, recruiting DNA exonucleases and other DNA modifying enzymes to mediate the V(D)J rearrangement of T and B cell antigen recognition receptor (BCR/Ig and TCR) genes. This process is crucial for the immune system to generate a vast number of B and T cell pools that specifically recognize different antigens.
The expression of RAG1 and RAG2 has strict tissue specificity and differentiation stage specificity, forming two peaks of expression during the differentiation and development of T and B cells. The first peak occurs during the progenitor T cell and progenitor B cell stages, mediating the rearrangement of BCR/Ig heavy chain and TCRβ chain genes. The second peak occurs during the pre-T and pre-B cell stages, mediating the rearrangement of BCR/Ig light chain and TCRα chain genes. Only when RAG molecules are effectively expressed can the V(D)J rearrangement of TCR genes and immunoglobulin light and heavy chain genes occur successfully, allowing T and B cells to continue differentiating and developing until maturity. Any nonsense or missense mutations in either the RAG1 or RAG2 gene resulting in abnormal expression of the RAG molecules will affect the activity of the recombination activating enzyme, leading to varying degrees of T and/or B cell development impairment, and severely affect the adaptive immune response function of the body. Therefore, the expression of RAG is closely related to the differentiation and development of lymphocytes.
Clinical Manifestations

RAG Complete Deficiency:
1) Typical SCID
Typical SCID is characterized by a complete loss of adaptive immune response function. Patients usually present symptoms within weeks after birth. The spectrum of pathogens is broad, primarily caused by infections from opportunistic pathogens affecting the lungs and intestines. Pathogens include fungi (Candida and Aspergillus), viruses (cytomegalovirus, parainfluenza virus, herpes simplex virus type I, varicella virus, respiratory syncytial virus, and adenovirus), and bacteria (disseminated Bacillus Calmette-Guérin, Pseudomonas). Bacterial infections are most commonly pneumonia, otitis media, and skin infections. Additionally, disseminated Bacillus Calmette-Guérin infections are also very common. The most common opportunistic virus is cytomegalovirus, which is an important marker of T cell deficiency. Without hematopoietic stem cell transplantation treatment, typical SCID patients often die within the first year after birth.
The immunophenotype of typical SCID is T−B−NK+, meaning patients cannot produce T and B cells but can produce NK cells. This severe lymphocyte deficiency is similar to other immunodeficiencies affecting V(D)J rearrangement function (Artemis, DNA ligase IV, Cernunnos). B cell deficiency leads to an inability to produce Ig, and eosinophilia may also occur.
Typical SCID is primarily characterized by abnormal development of immune organs and lymphoid tissues. Patients’ thymus development is severely affected, resulting in a very small thymus and a severe lack of thymocytes (T cells at different differentiation stages). Histopathological observations reveal significant changes: thymic lobules are separated by a thin layer of collagen, and immature thymocytes are scattered in a disordered arrangement within the thymic epithelial cell reticular structure. Patients’ thymus lacks the typical boundary features between the cortex and medulla and also lacks thymic corpuscles. However, thymic epithelial cells are normal and can regenerate thymocytes after hematopoietic stem cell transplantation. In peripheral immune organs, T cells are absent in the thymus-dependent areas of the spleen and lymph nodes. Mucosa-associated lymphoid tissues (such as tonsils, adenoids, and aggregated lymphoid nodules) are underdeveloped or completely absent.
2) Maternal T Cell Input Type SCID
Graft-versus-host disease (GVHD) mediated by maternal T cells is one of the important clinical features of maternal T cell input type SCID. GVHD most likely affects the skin, causing erythematous rash-like eczema that can easily be confused with allergic dermatitis, but the distinction is that in SCID patients, the affected areas are on the palms and soles, while allergic dermatitis can appear on any part of the skin. Other organs may also be affected by GVHD, with the liver and intestines being the most common. When the liver is involved, liver enzyme levels may increase, and eosinophilia and elevated IgE may occur. GVHD patients may also experience severe neutropenia, with granulocyte colony-stimulating factor treatment showing some efficacy. The severity of GVHD varies among individuals, and in fact, a certain proportion of patients do not develop GVHD.
The immunophenotype of maternal T cell input type SCID is T+B−NK+, which can easily be confused with Omenn syndrome (phenotype T+B−NK+), differing in the source of T cells. The T cells in maternal T cell input type SCID (carrying maternal HLA) are entirely of maternal origin, having crossed the placental barrier into the fetus during pregnancy, rather than being produced by the individual. The T cells are activated effector T cells (phenotype HLA DR+ CD45 RO+), but the number of cells varies greatly among different patients. Overall, the ratio of CD4+ T to CD8+ T cell subsets correlates with the severity of GVHD, with CD4+ T cells being predominant in severe GVHD patients, while mild and asymptomatic patients tend to have a predominance of CD8+ T cells, which may also be close in number to CD4+ T cells.
RAG Residual Deficiency:
1)Typical Omenn Syndrome (Omenn syndrome, OS)
OS can be divided into typical OS and atypical OS, with significantly different immunophenotypes and clinical presentations. Typical OS usually presents symptoms within weeks after birth, consistent with the characteristics of severe immunodeficiency disease. Similar to typical SCID, patients are prone to long-term diarrhea and severe recurrent infections. However, typical OS patients exhibit lymphadenopathy, hepatomegaly, and splenomegaly. The most prominent clinical manifestation is the appearance of significant diffuse erythroderma symptoms on the skin shortly after birth, especially with thickened fish-scale-like erythroderma.
Abnormal genes with RAG gene mutations are inherited in an autosomal recessive manner, primarily consisting of missense mutations, with occasional frameshift mutations. The above mutations belong to the category of hypomorphic allelic mutations in terms of phenotypic effects, meaning that patients can still express a certain residual activity of RAG molecules rather than complete deficiency.
The immunophenotype of typical OS is T+B−NK+. Residual RAG molecules are functionally impaired. Patients can still produce T lymphocytes in amounts below normal. Unlike maternal T cell input type RAG residual deficiency, the T cells are self-generated rather than maternal-derived fetal input cells. The T cell pool in patients mainly consists of oligoclonal αβ T cells, carrying only a limited diversity of αβ TCR molecules and unable to mediate effective immune protection, while the T cells in γδ T cell dominant RAG residual deficiency mainly consist of γδ T cells. The T cells in the blood are activated effector T cells (HLA DR+ CD45 RO+), and their main biological activity is primarily directed against self-tissues.
2)Atypical Omenn Syndrome
Patients usually present symptoms within weeks to months after birth. Symptoms such as growth failure, recurrent lung infections, and persistent diarrhea are similar to those seen in other types of RAG deficiency diseases. Erythroderma is relatively common, but most typical OS patients also exhibit similar symptoms. However, no reports of lymphadenopathy or splenomegaly have been found in atypical OS cases, which can serve as an important reference indicator for distinguishing atypical OS from typical OS and maternal T cell input types. Additionally, only a few patients exhibit hepatomegaly, whereas nearly all typical OS patients show this manifestation.
The immunophenotype of atypical OS is T+B+NK+. Unlike typical OS, a certain number of B cells and various Ig can be detected in peripheral blood, in addition to T cells. Furthermore, eosinophilia and elevated IgE are very significant.
3)Granulomatous Inflammation Type
Some patients have a very late onset. A typical case reported onset at age 3, with no prior infection history, while other RAG deficiency patients usually present symptoms within weeks to months after birth, showing a significant difference in onset time. The immunophenotype is T+B+NK+, with lymphocyte counts (0.32–1.6)×109/L, lower than that of normal children of the same age. Patients exhibit eosinophilia, but IgE does not elevate. T cell counts are significantly reduced [(0.1–1.1)×109/L]. The expression of effector T cell markers HLA DR and CD45 RO is enhanced. Maternal-derived T cells are undetectable, and there is no significant increase in T cells. CD4+CD45 RA+ cells (accounting for 1%–27% of CD4+ T cells) can reflect the presence of V(D)J rearrangement activity in the thymus and have the ability to produce naive T cells.
The most prominent histopathological feature of granulomatous inflammation type is the presence of epithelial-like, non-caseating granulomas in various organs and tissues (such as skin, lungs, tongue, tonsils, and spleen).
4)γδ T Cell Dominant Type (Expansion Type)
Patients experience severe viral infections within the first year of life, accompanied by autoimmune cytopenias. Specific antibodies against red blood cells, platelets, and neutrophils can be detected at this time. Most patients experience CMV infections, with occasional development into EBV infections associated with lymphoid tissue hyperplasia or genital ulcers caused by HSV 1 infection.
The immunophenotype of patients may be T+B+NK+ or T+B−NK+, with eosinophilia possible. Lymphocyte counts are below average normal levels. Unlike other RAG residual deficiencies, γδ T cell dominant type patients have γδ T cells accounting for 70%–90% of total T cells in peripheral blood, with counts reaching 3.2×109/L, although there are cases with counts as low as 0.3×109/L, predominantly composed of CD8+ γδ T cells.
5)Maternal T Cell Input Type
Case Sharing

A 3-month-old girl was hospitalized due to severe pneumonia, with a recent history of recurrent fever and malnutrition. Immunoglobulin tests showed IgG 0.47g/l, IgA<0.07g/l, IgM<0.04, IgE<5, NK94%, with total T0 and total B0, CD4/CD8=0, NK90%, and total cells 94%. Blood tests indicated: WBC 1.35, moderate 0.82<0.19. Clinically, there was a high suspicion of immunodeficiency-related disease.
Blood samples were sent to Kangxu Medical Testing Institute for immunodeficiency V2 detection package.
Testing found pathogenic or suspected pathogenic variations that could explain the patient’s phenotype.
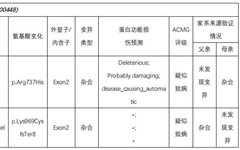
Selection Reason:
1. The following clinical phenotypes match the phenotype of the gene-related disease::Pneumonia (HP:0002090);Interstitial pneumonia (HP:0006515);Fever (HP:0001945);IgG antibody deficiency (HP:0004315);Severe combined immunodeficiency (HP:0004430); Combined immunodeficiency (HP:0005387)
2. Consistent with compound heterozygous nucleotide variations
ACMG Evidence: PS1, PM1, PP5, PP3 PVS1, PM2
Test Results:
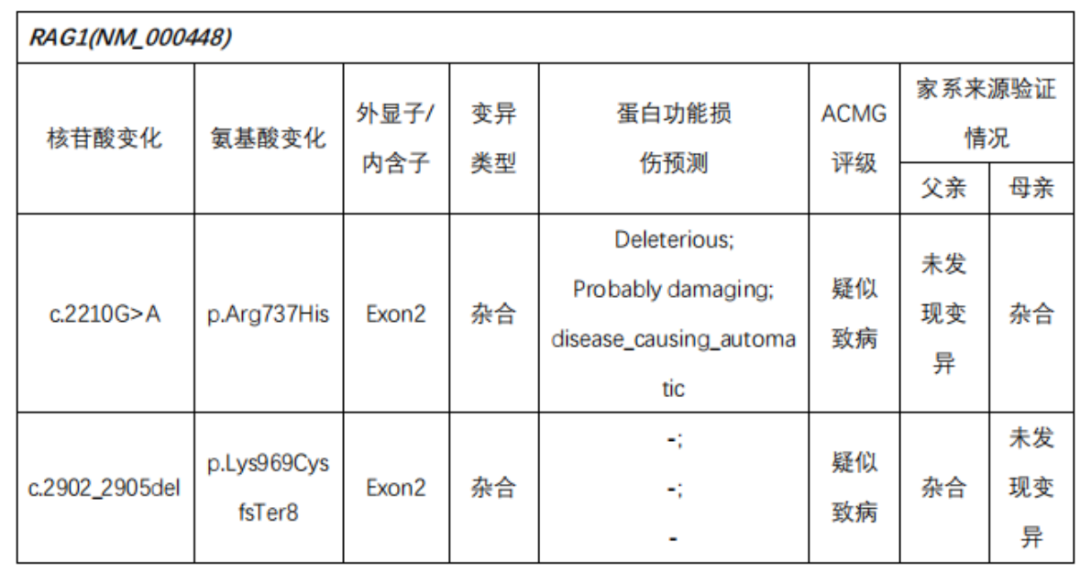
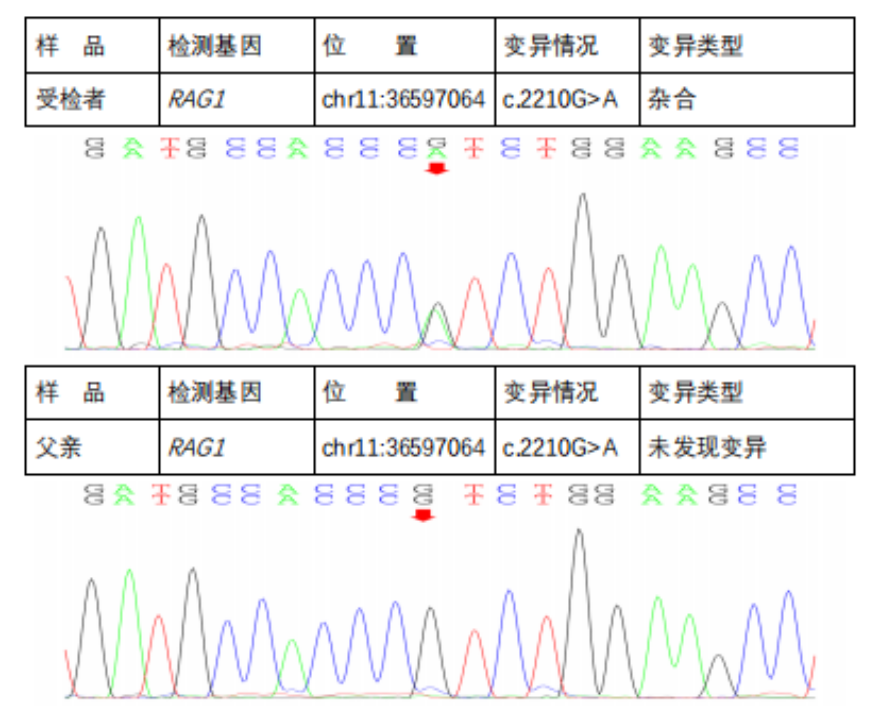
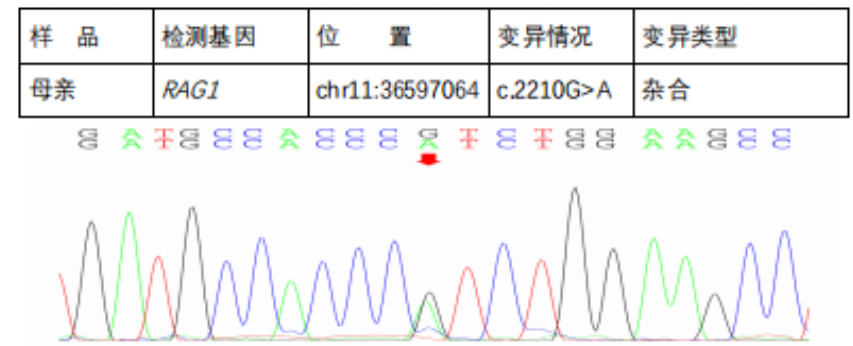
In the subject, a heterozygous nucleotide variation c.2210G>A (the 2210th nucleotide in the coding region changes from G to A) was found, which results in the change of the 737th amino acid from Arg to His (p.Arg737His), a missense mutation; a heterozygous nucleotide variation c.2902_2905del (deletion of nucleotides 2902_2905 in the coding region) was found, which leads to a change in amino acid synthesis starting from the 969th amino acid Lys, resulting in termination at the 8th amino acid after the change (p.Lys969CysfsTer8), a frameshift mutation. The above variations may affect protein function. The above variations were inherited from the parents, with each parent carrying only one of the heterozygous variations. The pathogenicity of variation c.2210G>A has been reported in the literature, reference: Partial V(D)J recombination activity leads to Omenn syndrome. PubMed: 9630231; the pathogenicity of variation c.2902_2905del has not been reported in the literature (reference databases: HGMD Pro and PubMed). The above variations do not belong to polymorphic changes and occur at a very low frequency in the population (reference databases: 1000Genomes, dbSNP). The RAG1 gene is a pathogenic gene for Omenn syndrome, severe combined immunodeficiency B cell (-) type, and combined cellular and humoral immune defects with granulomas, all inherited in an autosomal recessive manner (AR).The above variations may be pathogenic variations leading to the patient’s condition; further analysis and judgment combined with the patient’s clinical manifestations are recommended.
Clinical Symptoms of the Disease:
Omenn syndrome [autosomal recessive, failure to thrive, pneumonia, hepatomegaly, splenomegaly, diarrhea, systemic erythroderma; skin thickening, lymphocytic infiltration, with occasional histiocytic and eosinophilic infiltration, alopecia, anemia; thrombocytopenia; eosinophilia, lymphadenopathy; increased frequency of bacterial, viral, and fungal infections; significant changes in lymph node architecture: lack of follicles, lymphocyte depletion, increased proportion of reticular cells and eosinophils; extremely low B cells; B cell deficiency; increased numbers of activated CD45RO+, DR+ circulating T cells (usually increased); poor proliferative response of T lymphocytes to specific antigens; thymic hypoplasia, often lacking Hassall’s corpuscles; defects in the V(D)J recombination process, hypoproteinemia; extremely low IgG; extremely low IgA; extremely low IgM; increased IgE caused by mutations in recombination activating gene 1; caused by mutations in recombination activating gene 2].
Severe combined immunodeficiency, B cell (-) type [autosomal recessive, recurrent infections leading to growth failure, mastitis, otitis media, conjunctivitis, purulent rhinitis, recurrent acute pneumonia, diarrhea, arthritis, meningitis, frequent opportunistic infections; inability to reject allogeneic cells; peripheral blood B cell deficiency; peripheral blood T cell absence; significantly reduced or absent V(D)J recombination activity, hypogammaglobulinemia, symptoms appear at 2-3 months of age; if not treated promptly, death may occur within months, caused by mutations in recombination activating gene 1 (RAG1, {179615.0001)}; caused by mutations in recombination activating gene 2].
Combined cellular and humoral immune defects with granulomas [autosomal recessive, non-infectious granulomas; granulomas may appear in skin, tongue, lungs, or other tissues; hypogammaglobulinemia; ultrasound shows no thymus; reduced B cell count; reduced T cell count; T cell functional defects; residual effects of RAG1 and RAG2, onset in infancy or early childhood; T cell negative, B cell negative, NK cell negative SCID (601457) allele disease, symptoms are more severe, caused by mutations in recombination activating gene 1; caused by mutations in recombination activating gene 2].
Next-Generation Sequencing Validation Results::
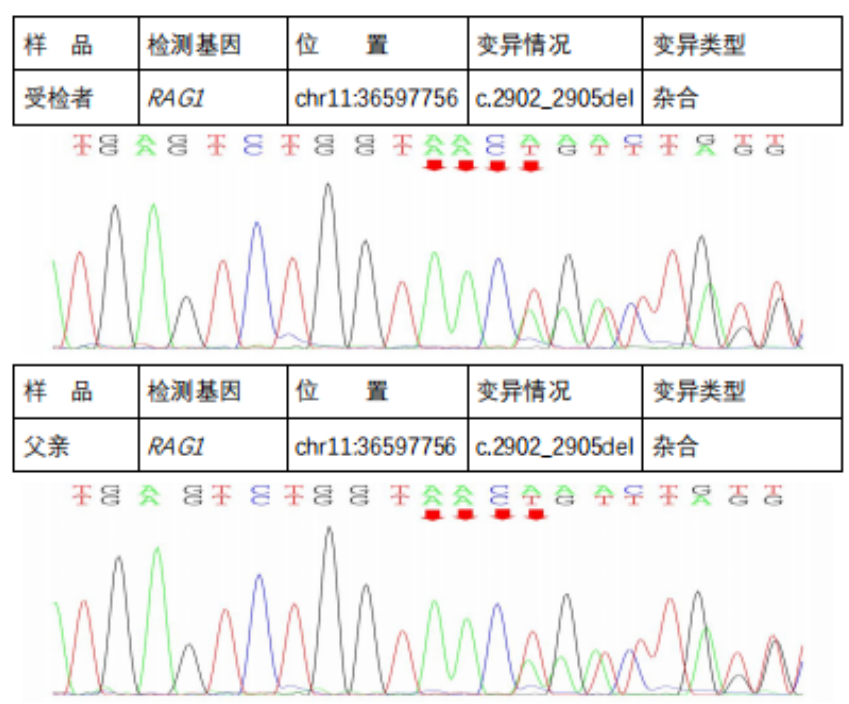
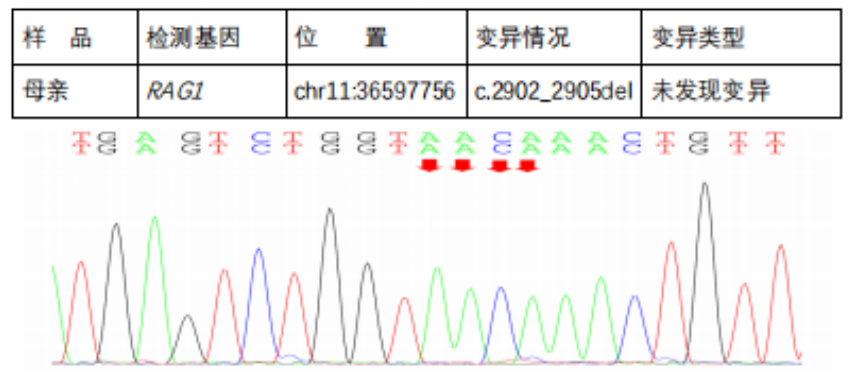
Diagnosis and Treatment

To treat SCID, immunoreconstruction must be performed through bone marrow transplantation (BMT) or gene replacement therapy to achieve survival. The highest success rate is achieved by using bone marrow tissue from HLA-matched siblings; using HLA-mismatched donor marrow tissue almost always leads to graft-versus-host disease and failure. Additionally, fetal liver or fetal thymus transplantation can be performed, but the efficacy is limited. Therefore, early diagnosis is crucial to enable timely immunoreconstruction and prevent the occurrence of difficult-to-treat infections that may threaten vital organs during the interval between diagnosis and immunoreconstruction.


【Case Sharing】 A Detection of Wolfram Syndrome Causing Recurrent Miscarriages
【Case Sharing】 Genetic Testing for Cerebellar Degeneration
【Case Sharing】 A Prenatal Diagnosis of Ectodermal Dysplasia
【Case Sharing】 Congenital Hypothyroidism Caused by DUOX2 Gene Mutation
【Case Sharing】 A Prenatal Diagnosis of Primary Glaucoma
【Case Sharing】 A Prenatal Diagnosis of Ectodermal Dysplasia
【Case Sharing】 5α-Reductase Type 2 Deficiency
【Case Sharing】 A Prenatal Diagnosis of 47, XYY Fetus with Supermale Syndrome
【Case Sharing】 A Prenatal Diagnosis of Chromosome Microdeletion at Xp22.33

