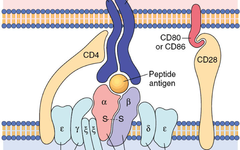



▉ Abstract
T cells receptors, co-stimulation signals, and cytokine signals coordinate the triggering of T cell activation and functional programming through specific signaling networks. Understanding immune metabolism signaling networks regulators and effectors may reveal therapeutic targets for modulating metabolic programs and T cell responses in human diseases.
In this article, we summarize the key signaling networks mediated by serine/threonine kinases, including the metabolic regulators of PI3K–AGC kinases, mTOR, and LKB1–AMPK pathways, particularly their regulatory roles in T cells. We focus on the cellular signaling pathways involved in immune metabolic regulation, primarily addressing molecules related to metabolic regulation, upstream and downstream targets, and their cell type-specific effects, categorized into mechanistic targets of PI3K, AGC, mTOR, and LKB1–AMPK signaling.
When T cell receptors (TCRs) recognize homologous antigens in the presence of co-stimulation and cytokine signals, the signaling network in naïve T cells is activated to promote clonal expansion, effector cell differentiation, and immune function. After antigen clearance, most effector cells die through programmed cell death, but some cells persist as memory cells, preparing for rapid recall in the next immune challenge. Moreover, the functional state of T cells has similar and distinct transcriptional programs, as well as differences in protein expression, activity, and interactions. Current research is just beginning to understand these differences, and there is increasing recognition of the importance of cellular metabolism in determining T cell development and function.
▉1. PI3K-AGC signaling
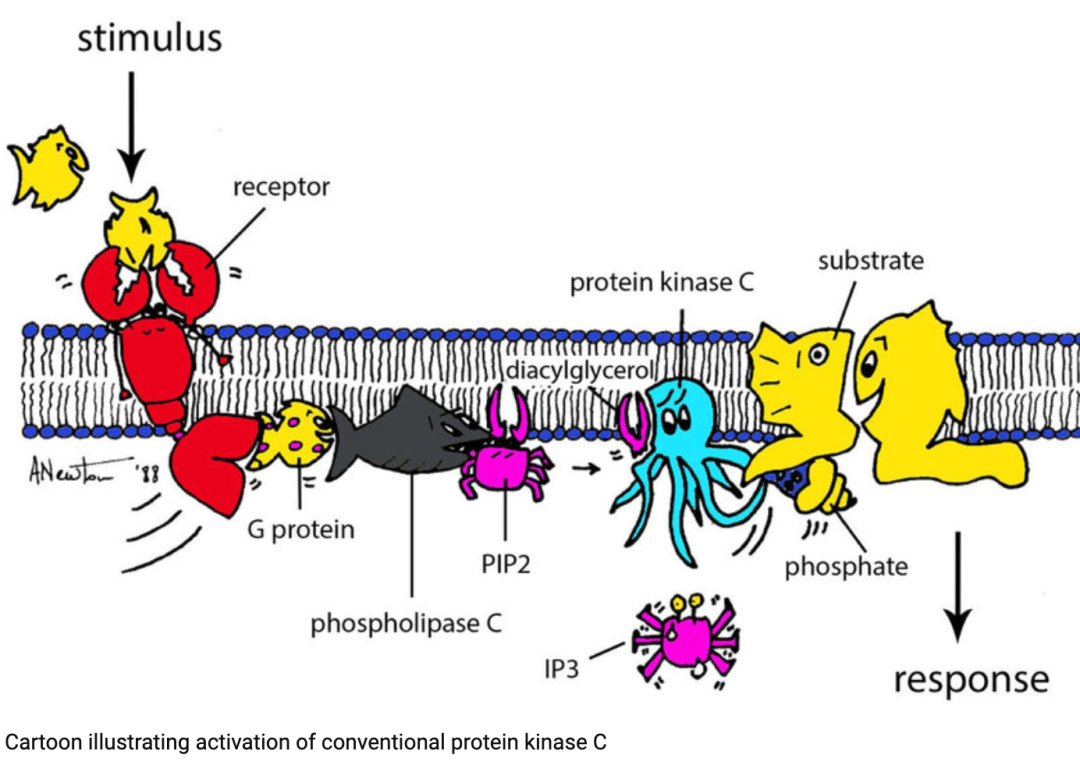
Phospholipids are key second messengers influencing downstream immune metabolic pathways, thus being highly regulated in conversion. A central mediator of phospholipid conversion is PI3K, which converts phosphatidylinositol- (4,5)-bisphosphate (PIP2) into phosphatidylinositol- (3,4,5)-trisphosphate (PIP3).PIP3 production leads to plasma membrane recruitment and functional regulation of proteins containing PH (pleckstrin homology) domains.
Thus, PI3K activity can generate subcellular signaling hubs by recruiting a large number of effector proteins containing PH domains to nearby areas. PI3K activity induces various signaling pathways involved in regulating cell function, including Akt (protein kinase B), phosphoinositide-dependent protein kinase 1 (PDK1), and mTOR complex 1.
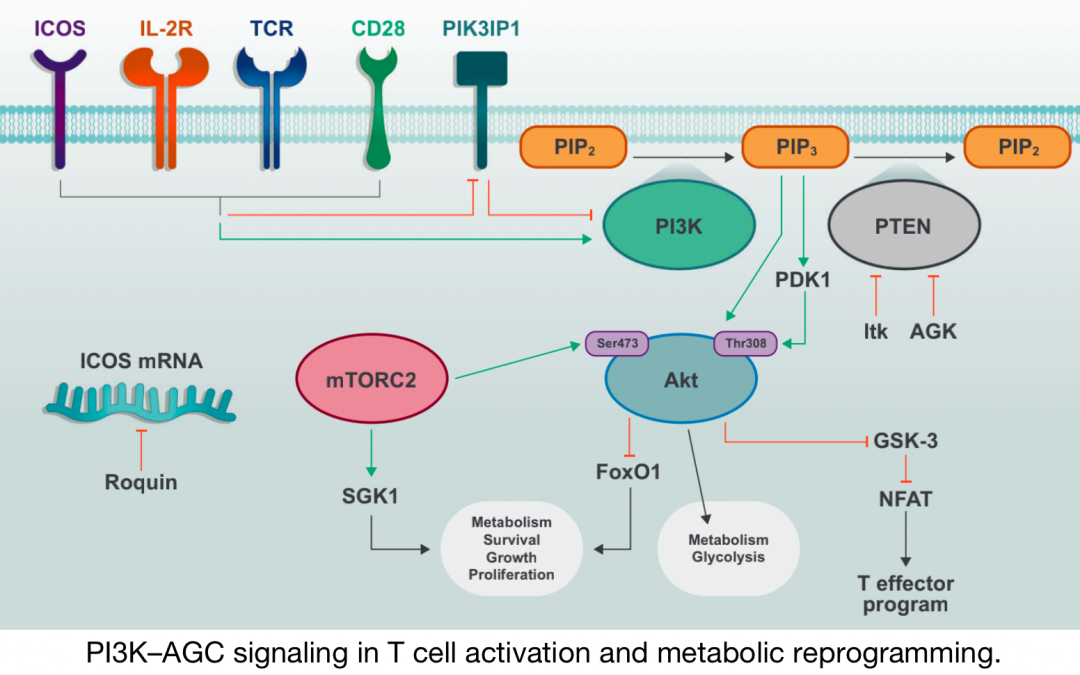
PI3K–T cell activation and metabolic reprogramming involve AGC signaling
Activation of TCR, CD28, and IL-2R induces phosphorylation and activation of PI3K, as well as inactivation of PI3K inhibitory molecules. PIP2 is converted to PIP3 through the activity of PI3K, and PIP3 facilitates the plasma membrane recruitment and activation of downstream signaling molecules including PDK1 and Akt. mTORC2 further activates Akt and promotes metabolic increases and T cell effector functions.
▉1.1PI3K
《Protein kinase C: perfectly balanced》
Class I PI3K is a heterodimeric protein, consisting of one catalytic p110 subunit (p110α, p110β, p110δ, or p110γ) and one of five regulatory subunit isoforms (p50α, p55α, p55γ, p85α, or p85β). In the absence of stimulation, the kinase domain of the catalytic subunit is inhibited by the regulatory subunit. The concentration of PI3K substrates is closely related to cell activation status; in T cells, cell activation status is determined by TCR binding, CD28 family-mediated co-stimulation, and cytokine signals (such as downstream of IL-2R). Upon upstream activation, the Src-homology-2 (SH2) domain of PI3K can bind to specific phosphorylated YXXM motifs on upstream receptors or adaptor proteins, leading to the release of the inhibitory interaction of the regulatory subunit and translocation of the catalytic subunit to the substrate-rich plasma membrane.
PI3K signaling is negatively regulated by phosphatase activity. Specifically, PIP3 is converted to PIP2 or PI-(3,4)-P2 by phosphatases and tension protein homologue (PTEN), as well as by inositol 5′-phosphatases (SHIP) containing SH2 domains. SHIP plays a crucial role in promoting Th1 cell responses, while PTEN deficiency leads to T cell hyperactivation, especially under suboptimal stimulation. PTEN is inhibited under strong TCR stimulation or CD28 co-stimulation, partly due to Tec family kinase IL-2-induced T cell kinase (Itk), and is also negatively regulated by AGK-mediated phosphorylation during CD8 T cell activation.
▉1.2 AGC Kinases
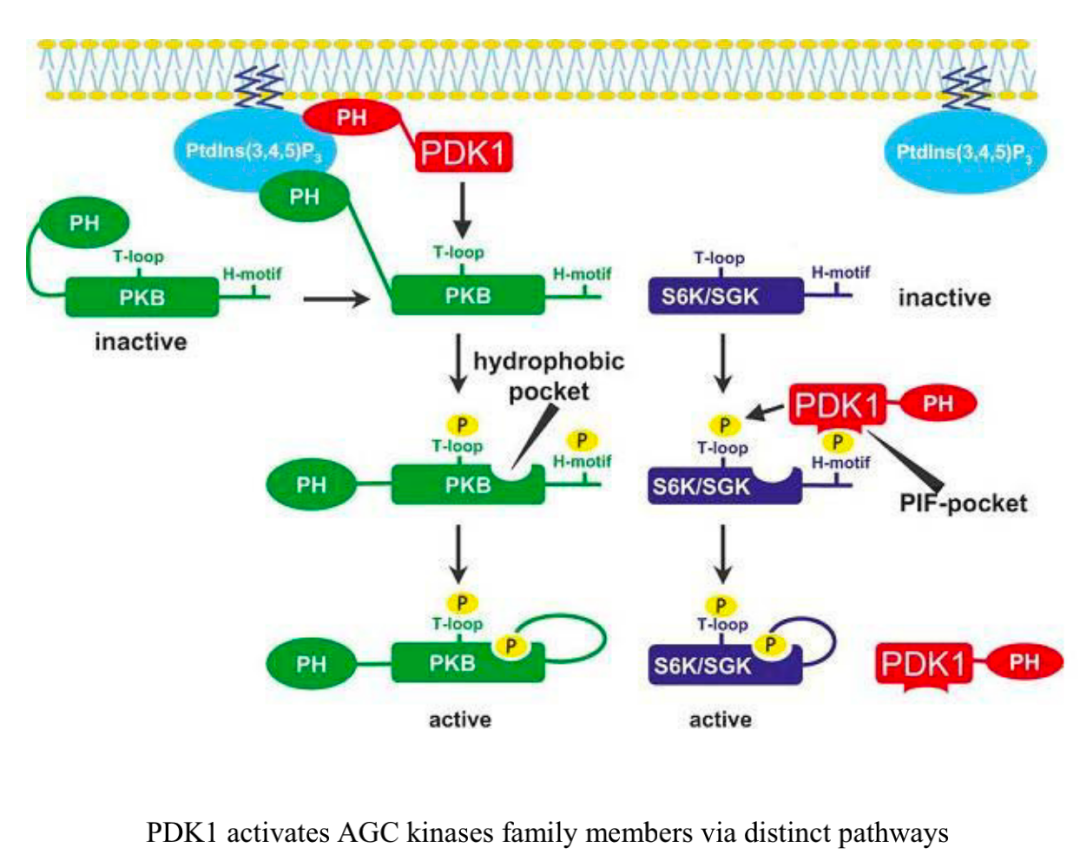
Among the most prominent proteins containing PH domains are members of AGC kinases, including PDK1, Akt, ribosomal S6 kinase (RSK, also known as p90) and serum/glucocorticoid-regulated kinase 1 (SGK1).PDK1 is a serine/threonine kinase that plays a crucial role in activating other AGC kinases (including Akt and SGK1). Activation of TCR and CD28 promotes recruitment of PDK1 to the plasma membrane and induces its phosphorylation, specifically through protein kinase C-θ (PKC-θ) dependent phosphorylation at Thr513, rather than autophosphorylation of the kinase domain residue (Ser241), which is critical for driving T cell activation. In activated CD8 T cells, PDK1 is involved in metabolic reprogramming, maintaining glucose uptake and glycolysis downstream of IL-2 stimulation, a process that requires mTOR–HIF-1α (hypoxia-inducible factor-1α) axis, but does not require PI3K or Akt activity. Proper regulation of PDK1 activity is crucial for enhancing appropriate T cell activation and controlling Treg cell function in inflammation. After phosphorylation at Ser473 by mTORC2, Akt reaches maximal activation, which allowsAkt to bind to a substrate dock known as the PDK1 “PIF pocket”, facilitating phosphorylation of Akt at Thr308.mTORC2–Akt signaling can then coordinate T cell activation, differentiation, and trafficking.
《The Clinical Implications of the Survival Pathway in Prostate Cancer》
One important role of Akt in T cells is to regulate the activity of forkhead box O (FoxO) transcription factors through phosphorylation, leading to their exclusion from the nucleus and termination of target gene transcription.FoxO proteins are more transcriptionally active in resting cell populations, promoting the expression of the homeostatic cytokine receptor IL7R and cell trafficking molecules (such as CD62L, CCR7, and S1PR1) through KLF2. Recent evidence suggests that proper downregulation of FoxO1 activity is crucial for T cell intracellular environment stability. Mechanistically, downregulation of FoxO1 in activated T cells is important for coordinating cell growth and proliferation, as it allows for sustained mTORC1 signaling and anabolic metabolism; thus, the Akt–FoxO1 axis dynamically regulates T cell responses.
Another serine/threonine kinase involved in Akt signaling is glycogen synthase kinase 3 (GSK-3), which has constitutive activity in resting cells, but is inactivated by phosphorylation through Akt. Active GSK-3 promotes the survival of activated T cells by limiting the activity of nuclear factor (NFAT) in T and B cells and restricting activation. The exact roles of AGC kinases (such as RSK) and their substrates in T cell metabolic programming have been a focus of attention and are also a current research priority.
▉2. mTOR signaling
mTOR is a serine/threonine protein kinase that exists in two signaling complexes mTORC1 and mTORC2 . mTORC1 is defined by the mTOR kinase, and its adaptor proteins regulate mammalian lethal proteins related to mTORC1 and the proline-rich Akt substrate-1 (PRAS1) and its target DEP domain-interacting protein (DEPTOR) inhibitory subunits. mTORC1 signaling is crucial for T cell development in the thymus, peripheral homeostasis, and differentiation into effector CD4 Th1, Th2, and Th17 cells, as well as cytotoxic CD8 T cells. In contrast, Th1 and Th2 cell differentiation requires mTORC2 activity, which can also regulate the migration of Tfh and Treg cells.
mTOR signaling plays an additional role in antagonizing traditional T cell responses by regulating the activation, lineage stability, and suppressive functions of Treg cells. Finally, mTOR signaling can antagonize the differentiation of long-lived memory CD8 T cells in lymphoid tissues, while promoting their development in non-lymphoid tissues. Thus, mTOR signaling serves as a central regulator of T cell responses.
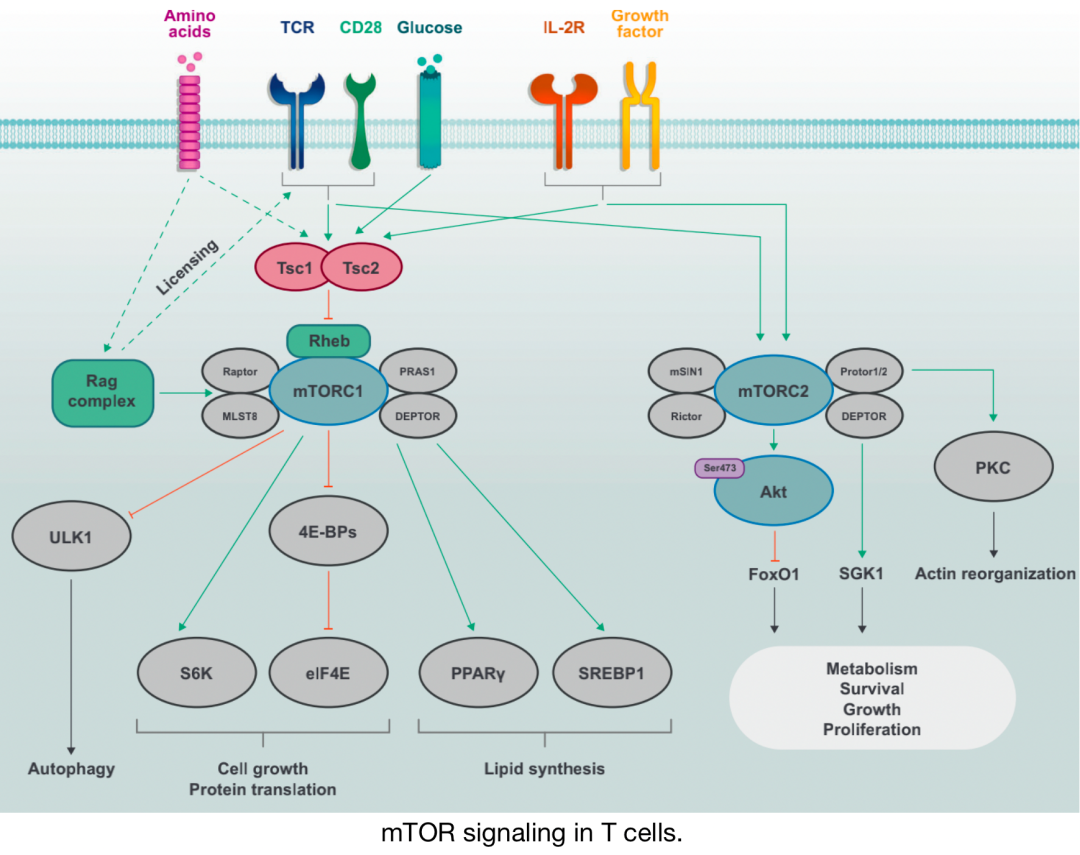
mTOR signaling in T cells
Discrete mTOR complexes mTORC1 (composed of mTOR, Raptor, PRAS1, DEPTOR, and MLST8) and mTORC2 (composed of mTOR, Rictor, Protor1/2, mSIN1, and DEPTOR) are activated by immune receptors (TCR, CD28, and IL-2R) and growth factors. Activation of mTORC1 is also sensitive to nutrients such as amino acids, which promote mTORC1 activation through the Rag complex; the Rag complex also plays a permissive role in allowing TCR and CD28 co-stimulation signals to induce mTORC1 activation. The activity of the Tsc complex is inhibited by immune and growth factor signals by inhibiting the activation of the small G protein Rheb, which promotes mTORC1 activation. mTORC1 induces cell growth and protein translation through S6K and eIF4E and inhibits autophagy through ULK1 under nutrient-rich conditions. mTORC2 primarily plays a critical role in cell growth, proliferation, and survival, or sub-specific metabolic reprogramming.
▉3. LKB1-AMPK signaling
LKB1–AMPK signaling pathways play a core role in regulating cell metabolism, proliferation, and survival in response to changes in nutrient and energy demands. LKB1–AMPK signaling can promote catabolic pathways that generate ATP and enable T cells to have metabolic plasticity in response to energy stress. By regulating metabolic reprogramming, LKB1 and AMPK contribute to the differentiation and function of T cells. In the following sections, we will discuss how LKB1 and AMPK activity is regulated, their impact on metabolism, and their role in T cell-mediated immunity.
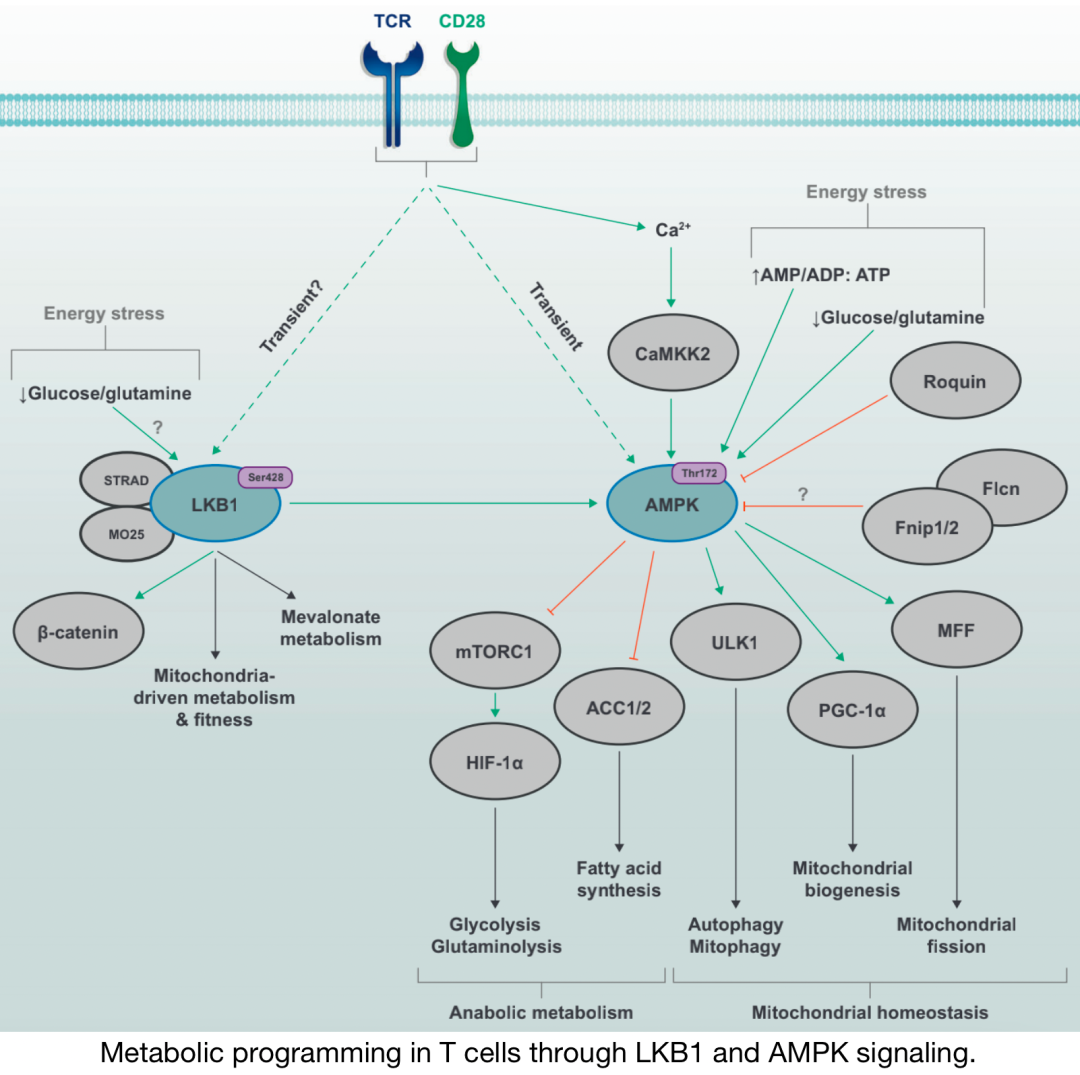
Metabolic programming in T cells through LKB1 and AMPK signaling
The energy stress pathway kinases LKB1 and AMPK are activated by TCR and CD28 co-stimulation signals; AMPK activity is partly mediated by Ca2+CAMMK2 pathways. In conditions of glucose or glutamine deficiency or imbalance of AMP/ADP/ATP ratios, it can also promote LKB1–AMPK signaling. Upstream nutrient-sensing proteins, such as the Fnip–Flcn complex and Roquin, can inhibit AMPK function. Activation of LKB1 is associated with changes in mitochondrial metabolism and fitness, as well as increased mevalonate metabolism in specific environments. By regulating the activity of multiple downstream targets, AMPK signaling can block metabolic programs related to glycolysis, glutaminolysis, and fatty acid synthesis while promoting catabolic processes, such as mitophagy and autophagy. AMPK also drives mitochondrial biogenesis and dynamics by promoting mitochondrial apoptosis, thus supporting mitochondrial activity.
▉3.1 Regulation of LKB1 and AMPK
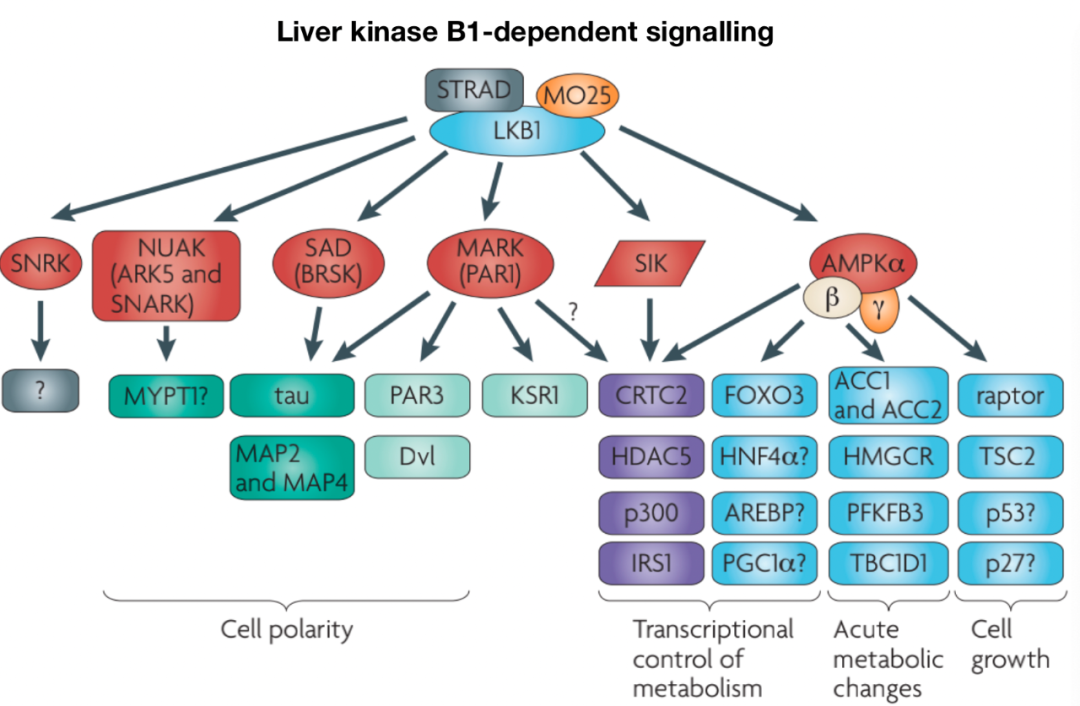
《The LKB1-AMPK pathway: metabolism and growth control in tumour suppression》
LKB1 is a serine/threonine kinase with tumor suppressor functions involved in regulating cell metabolism and proliferation. LKB1 has many downstream targets, including the most clearly defined targets AMPK and AMPK-related kinases, such as BRSK, NUAK, and MARK. The upstream regulation of LKB1 is mediated by cellular localization and post-translational modifications. LKB1 forms a complex with STE20-related adapter (STRAD) and MO25, promoting LKB1’s cytoplasmic localization and kinase activity. Additionally, Akt can inhibit LKB1 by promoting its nuclear retention.
AMPK is a conserved serine/threonine kinase composed of α, β, and γ subunits. Binding of AMP and ADP to CBS3 repeat sequences restricts phosphatases’ access to Thr172 on the AMPK catalytic α subunit, which is the key phosphorylation site that promotes AMPK kinase activity. Ultimately, complex regulation of metabolic products allows AMPK activity to respond in a “graded” manner under different energy stress conditions. At least three upstream kinases mediate AMPK Thr172 phosphorylation: LKB1, calcium/calmodulin-dependent kinase kinase 2 (CaMKK2), and TGF-β-activated kinase 1 (TAK1), with LKB1 and CaMKK2 playing the most clear roles. In T cells, TCR and Ca2+ signals and CD28 co-stimulation synergistically activate AMPK rapidly and transiently through CaMKK2. AMPK activity is also regulated through protein-protein interactions. Fnip1 and Fnip2 form complexes with AMPK, and loss-of-function mutations in Fnip1 enhance AMPK activity in B cells, establishing Fnip1 as a negative regulator of AMPK. The Fnip-binding protein Flcn also interacts with AMPK and may serve as a negative regulator of AMPK signaling, with evidence suggesting that Fnip1 and Flcn are reciprocally regulated by AMPK.
▉ 3.2 LKBI-AMPK Mediated Catabolism
AMPK activity is associated with low nutrient abundance induction, typically related to catabolic programs for generating ATP. AMPK activates autophagy through phosphorylation of ULK1, which promotes autophagy-mediated mitochondrial homeostasis, enhancing T cell survival. Additionally, AMPK regulates independently of mTOR; under glucose-limited conditions, AMPK also mediates cell cycle arrest through phosphorylation of p53 at Ser15. Therefore, LKB1–AMPK signaling promotes cell survival by limiting anabolic metabolism and cell growth during energy stress. Furthermore, metabolites produced through the pentose phosphate pathway, such as ribulose-5-phosphate, promote the disruption of the LKB1–AMPK pathway, facilitating lipogenesis. Conversely, LKB1 can also promote mevalonate metabolism to support Treg cell homeostasis in an AMPK-independent manner. Thus, LKB1–AMPK signaling primarily acts to inhibit lipid synthesis pathways, but AMPK-dependent LKB1 signaling may promote de novo lipogenesis under selective circumstances.
Studies on tumor cells indicate that disruption of LKB1 or AMPK signaling promotes aerobic glycolysis, partly through HIF-1α, thus increasing transcription of glycolytic enzymes. LKB1–AMPK-dependent regulation of HIF-1α may also partly depend on inhibition of mTORC1. LKB1–AMPK signaling may indirectly coordinate differentiation of Th17 and Treg cell lineages through changes in glycolysis and mitochondrial oxidative metabolism mediated by HIF-1α or ACC1. Additionally, recent studies suggest that LKB1 promotes stable Foxp3 expression and development of Th2-like Treg cells, independent of AMPK and mTORC1–HIF-1α signaling, but reliant on β-catenin signaling. LKB1 signaling is essential for mitochondrial function and mitochondrial-dependent metabolic programs following TCR-mediated Treg cell activation, including FAO or purine and pyrimidine metabolism. These findings emphasize the coordination of LKB1 and AMPK in metabolic reprogramming to regulate T cell differentiation and Treg cell function.
▉4. Immune Metabolism Signaling Networks
The major signaling pathways discussed above are not functionally exclusive; rather, they intertwine and interact to achieve appropriate metabolic regulation to meet the specific environmental demands of cellular functions. Thus, many of these regulatory inputs converge at common nodes, including individual molecules and entire cellular processes.
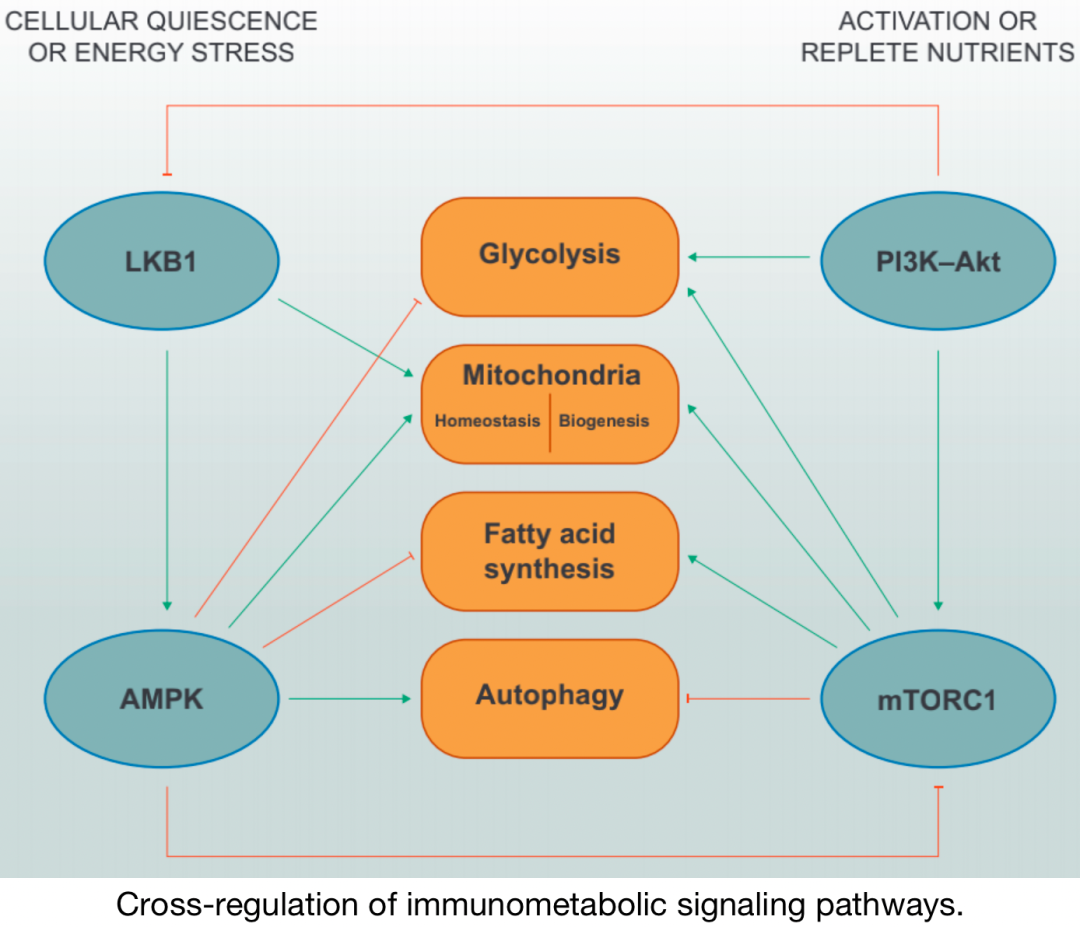
Cross-regulation of immune metabolic pathways
Under nutrient-deprived conditions, LKB1–AMPK signaling inhibits anabolic metabolic programs such as glycolysis and fatty acid synthesis while promoting mitochondrial homeostasis and autophagy. AMPK directly inhibits mTORC1 through phosphorylation of its specific adaptor protein Raptor. Under activated and/or nutrient-rich conditions, PI3K–Akt and mTORC1 signaling promotes glycolysis, mitochondrial biogenesis, and fatty acid synthesis, while inhibiting autophagy. It has been reported that Akt can phosphorylate LKB1 to inhibit its functional localization.
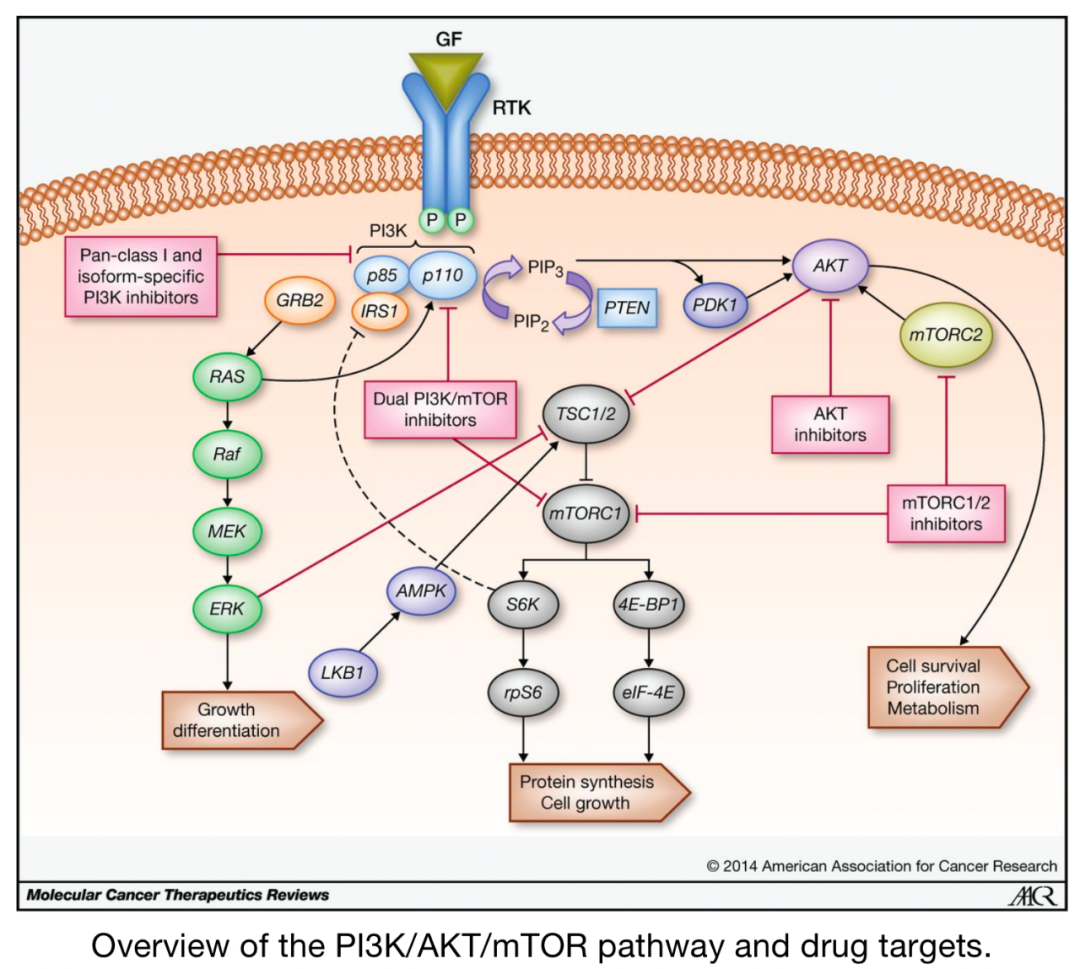
In general, increased activity of PI3K–AGC kinases and mTOR, along with decreased activity of LKB1–AMPK, is associated with cell growth, proliferation, and effector functions, which correlates with changes in metabolic programs. The signaling integration of upstream complexes ultimately relates to the regulation of downstream metabolic programs, determining the signaling pathways of immune cell metabolic programs, which are also influenced by interactions with metabolites and nutrients. This “bidirectional metabolic signaling” provides another layer of regulation for kinase-dependent signaling and gene transcription to control immune cell functions in different environments.
The role of immune signaling networks in immune metabolism is an exciting research area with broad implications for therapy and human health. In this review, we emphasize the key roles of PI3K–AGC, mTOR, and LKB1–AMPK signaling in T cell function and fate. This field has made significant progress in understanding how these signaling molecules are regulated and how they reprogram T cell metabolism, identifying relatively well-known upstream immune signals (such as TCR and cytokine receptors) and how they transmit different functional programs in metabolism, which may provide new therapeutic targets for improving adoptive T cell therapy (such as anti-tumor CAR-T cell therapy).
References:Saravia, J., Raynor, J.L., Chapman, N.M. et al. Signaling networks in immunometabolism. Cell Res 30, 328–342 (2020). https://doi.org/10.1038/s41422-020-0301-1
Editor: Ning Shuai
This post is intended for knowledge dissemination. For any copyright concerns, please contact us within 30 days of publication.
Unauthorized reproduction of original content on other platforms is prohibited.
If you have questions, please email [email protected] for more information.
©2021 Medical Overview All rights reserved
Previous Links“The Journey of the Little Vaccine” | Overview of Pharmaceutical Companies’ Pipelines Everyone Understands Immunology | Everyone Understands Immunology (Audio Version) Review Article Interpretation | Literature Briefing | Medical Popular Science | Pharmaceutical Frontier NotesPROTAC Technology | Antibody Drugs | Antibody Drug Conjugates – ADC Nucleic Acid Vaccines | CAR Technology | Chemical Biology

Warm Reminder
 The Medical Overview public account currently has nearly 7 discussion groups (enthusiastic, interesting, and gathering talents in the pharmaceutical circle). To join the group, add the author’s WeChat (xs2014233) or scan the public account QR code to add the author, noting “Name/Nickname – Company/University – Specific Research Field/Major”. This group is only for academic discussions, please do not disturb. Simply operate to star⭐️ Medical Overview, to receive our push notifications promptly① Click on the title below “Medical Overview” ② Go to the upper right corner “…” ③ Click “Set as Star”
The Medical Overview public account currently has nearly 7 discussion groups (enthusiastic, interesting, and gathering talents in the pharmaceutical circle). To join the group, add the author’s WeChat (xs2014233) or scan the public account QR code to add the author, noting “Name/Nickname – Company/University – Specific Research Field/Major”. This group is only for academic discussions, please do not disturb. Simply operate to star⭐️ Medical Overview, to receive our push notifications promptly① Click on the title below “Medical Overview” ② Go to the upper right corner “…” ③ Click “Set as Star”