

Single-cell RNA sequencing (scRNA-seq) technology enables gene expression detection across the transcriptome within individual cells, which can be used to study somatic clonal structures and characterize cellular heterogeneity in complex diseases. However, the data from scRNA-seq analysis are characterized by complexity, uncertain distributions, large data volumes, and high missing rates, making scRNA-seq analysis for biological inference challenging. Previous studies, such as Phenograph3, MAGIC4, and Seurat5, used k-nearest neighbors (KNN) algorithms to simulate relationships between cells. However, this may overly simplify the complex relationships between cell populations and genes.
In recent years, researchers have drawn on ideas from convolutional networks, recurrent networks, and deep autoencoders to define and design neural network structures for processing graph data—Graph Neural Networks (GNNs). GNN is a connection model that decouples node relationships in graphs by propagating adjacent information within deep learning architectures. Unlike other autoencoders used in scRNA-seq analysis, the unique aspect of graph autoencoders is their ability to learn low-dimensional representations of graph topology and train node relationships from a global view of the entire graph.
Recently, researchers from the University of Missouri’s Department of Electrical Engineering and Computer Science, the Ohio State University College of Medicine’s Department of Biomedical Informatics, and the Department of Neuroscience innovatively introduced a multimodal framework based on Graph Neural Networks (GNN)—Single-cell Graph Neural Network (scGNN)—to simulate the heterogeneous intercellular relationships and their potential complex gene expression patterns from scRNA-seq, providing a hypothesis-free deep learning framework for scRNA-seq analysis. scGNN integrates three iterative multimodal autoencoders and outperforms existing gene imputation and cell clustering tools across four benchmark scRNA-seq datasets. Furthermore, in a study of Alzheimer’s disease, scGNN successfully analyzed neurodevelopmental factors and potential regulatory mechanisms associated with the disease. The research findings have been published in Nature Communications, titled “scGNN is a novel graph neural network framework for single-cell RNA-Seq analyses.”
The article was published in Nature Communications.
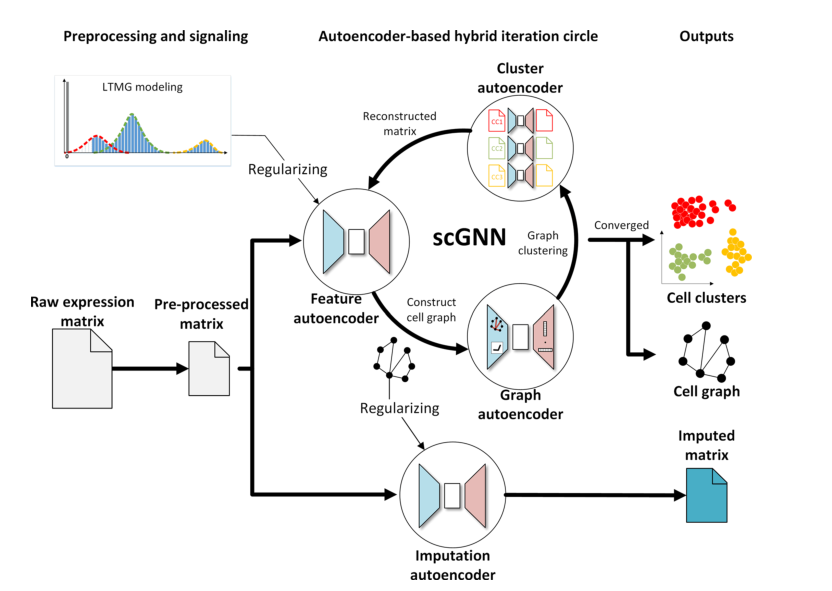
scGNN was developed to identify effective cellular and gene expressions. The main structure of this framework consists of three iterative autoencoders, including feature autoencoder, graph autoencoder, and clustering autoencoder. scGNN takes the gene expression matrix generated from scRNA-seq as input, uses LTMG (Left-Truncated Mixture of Gaussians) to regularize the input gene expression data, and converts it into discretized regulatory signals. The feature autoencoder learns low-dimensional embeddings by reconstructing gene expression and regularization, exploring deep features of cell data, and constructs and prunes a cell graph based on this. The graph autoencoder takes the processed cell graph as input and learns the topological embeddings of the cell graph for cell clustering. The clustering autoencoder reconstructs the expression matrix of the feature autoencoder in each cell cluster. Based on the cell type information inferred by the graph autoencoder, the clustering autoencoder can handle different cell types and regenerate expressions within the same cell cluster, aiding in discovering specific cell type information in personalized learning for each cell type.
Gene imputation aims to address the widespread missingness in scRNA-seq data, where a significant number of actively expressed genes are marked as zero. Existing imputation methods, such as MAGIC and SAVER, still face challenges in estimating differential gene expression and tend to produce false positives and biased gene correlations. To validate the gene imputation performance of scGNN, four benchmark scRNA-seq datasets were selected, and scGNN was compared with nine existing gene imputation tools, revealing that scGNN excels in restoring gene expression. Meanwhile, researchers found that scGNN can recover potential gene-gene relationships omitted due to the sparsity of scRNA-seq in the original expression data, capturing true gene-gene relationships and enhancing DEG (Differentially Expressed Gene) signals without introducing additional noise. Furthermore, researchers evaluated the clustering performance of scGNN and nine imputation tools on two identical datasets. The results showed that compared to the other nine imputation tools, when using scGNN embeddings, the distinctions between color blocks were more pronounced, and cells within the same cluster grouped more closely, while different clusters were more separated.

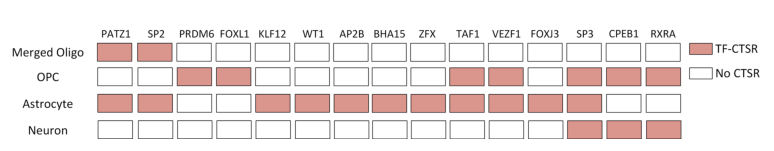
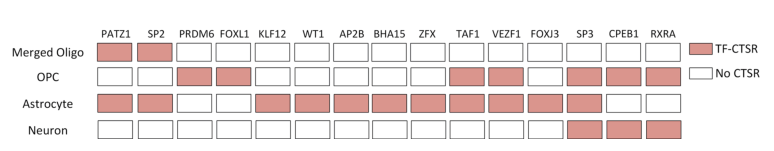
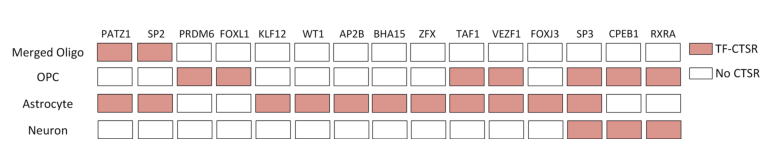
To further demonstrate the application capability of scGNN, researchers validated it using data from Alzheimer’s disease research, which included 13214 scRNA-seq datasets collected from six AD and six healthy control brains. The gene expression matrix generated from scRNA-seq was used as input for IRIS3, identifying 21 cell type-specific regulatory subunits (CTSR) across five cell types. The analysis discovered several transcription factors (TFs) and target genes associated with AD, which have been reported to be involved in the development of AD. Additionally, the SP3 factor, which can regulate neuronal synaptic functions, was found across all cell groups and was highly activated in AD. CTSRs regulated by the SP3 factor were found in OPCs, Astrocytes, and Neurons, indicating significant changes in SP3-related regulation within these three cell groups. These findings provide direction for discovering SP3 factors in Alzheimer’s disease research.
Exploring cellular heterogeneity in high-capacity, high-sparsity, and noisy scRNA-seq data remains a fundamental challenge. Existing studies indicate that the higher-order topological relationships of the complete cell graph have not been well explored and expressed. The key innovation of scGNN in this study is the integration of global propagation topological features of cells through GNN, along with the integration of gene regulatory signals during the iterative process of scRNA-seq data analysis. scGNN provides effective representations of gene expression and intercellular relationships, serving as a powerful learning framework applicable to general scRNA-seq analysis.
· END
 Popular ArticlesRecommended
Popular ArticlesRecommended
Nature | MD Anderson Cancer Center Develops Novel Single-Cell Sequencing Method to Confirm Subclonal Diversity in Breast Tumors During Progression
Nature Medicine | Cambridge University Team Develops Semi-Automated Classification Model Based on Deep Learning from Pathological Images to Assist Early Detection of Esophageal Cancer Precursors
JCI | He Jianxing/Fan Jianbing and Team Jointly Develop Blood ctDNA Methylation Model, Expected to Become a Powerful Tool for Non-Invasive Diagnosis and Typing of Lung Nodules
PNAS | Jiang Rui/Wang Yongxiong Team Publishes Deep Generative Neural Network Method for Probability Density Estimation

Don’t forget to click “Look” if you like it!