Compiled by | Jiang Changzhi
Today, I would like to introduce an article published in Nature Methods by a team from ETH Zurich and Friedrich Schiller University Jena. The article proposes a new method for generating small molecule structures from mass spectrometry based on an encoder-decoder neural network: MSNovelist. It first uses SIRIUS and CSI: FingerID to predict the molecular fingerprints and expressions from mass spectrometry, which are then input into an encoder-decoder based RNN model to generate the SMILES of the molecules. The authors evaluated MSNovelist using a dataset of 3863 mass spectrometry data from the Global Natural Product Social Molecular Networking (GNPS) website, where MSNovelist reproduced 61% of the molecular structures, all of which were unseen in the training set; it also achieved a reproduction rate of 64% using the CASMI2016 dataset. Finally, the article validated MSNovelist on a dataset of moss plant mass spectrometry data, showing that MSNovelist is very suitable for annotating molecules corresponding to mass spectrometry in cases where the analytes and new compounds perform poorly.

Research Background
The identification of small molecules is an important task in life sciences, and mass spectrometry can be used to analyze compound components. Therefore, obtaining small molecule structures by analyzing mass spectrometry data is of great significance. Currently, there are spectral libraries containing tens of thousands of annotated small molecules, paving the way for machine learning and even deep learning methods to identify small molecule structures from mass spectrometry data. Existing methods are mostly based on mass spectrometry database matching and searching, which cannot identify new compounds, such as unknown natural products, drug metabolites, or environmental transformation products. In principle, the simplest method to identify the structural identity of unknown compounds, completely independent of the mass spectrometry database, is to first determine the molecular formula, then enumerate all possible candidates, and finally score them based on real data. However, this method faces the combinatorial explosion problem of structural quantities. On the other hand, deep learning models used for targeted de novo molecule generation do not have this problem and can query a vast chemical space of new compounds. Therefore, generative deep learning models hold great potential for the task of identifying new compounds from mass spectrometry.
2
Main Contributions
(1) This article proposes an encoder-decoder based RNN model to generate the corresponding SMILES of molecules from mass spectrometry data, named MSNovelist. MSNovelist first uses SIRIUS and CSI: FingerID to predict the molecular fingerprints and expressions from mass spectrometry, which are then input into an encoder-decoder based RNN model to generate the SMILES of the molecules;
(2) This article validates MSNovelist on a moss plant mass spectrometry dataset, and the experimental results show that MSNovelist is very suitable for annotating molecules corresponding to mass spectrometry in cases where analyte categories and new compounds perform poorly.
3
Model
3.1 Data Preprocessing
The dataset used in this article consists of data from HMDB (4.0), COCONUT, and DSSTox databases. The molecules in the training set were filtered to remove those that could not be parsed by RDKIT, those with SMILES exceeding 127 characters, those containing split points in SMILES, those with a molecular weight greater than 1000 Da, and those with more than 7 rings (as specified in SMILES) or elements other than C, H, N, O, P, S, Br, Cl, I, and F. Finally, the training set contains 1,232,184 molecules, which have 1,048,512 different structures (according to InChIKey2D rules).
3.2 Method
MSNovelist model inputs mass spectrometry data to generate its molecular structure based on the mass spectrometry information, predicting what molecular compound corresponds to the mass spectrometry. First, MSNovelist uses SIRIUS and CSI: FingerID to predict the molecular expressions and structural fingerprints from mass spectrometry, where the structural fingerprint is a 3609-dimensional vector representing the possible molecular structural features of the mass spectrometry; then, the molecular expression and structural fingerprint are input into an encoder-decoder RNN model, which can generate the SMILES expression of the molecule from scratch based on the fingerprint feature vector under the constraint of the molecular expression; finally, the modified Platt score is used to calculate the score between the generated molecule and the real mass spectrometry fingerprint as a loss to optimize the model parameters. The encoder of the RNN model consists of 3 hidden layers, and the decoder is an LSTM with 3 layers. The framework of the MSNovelist model is shown in Figure 1.
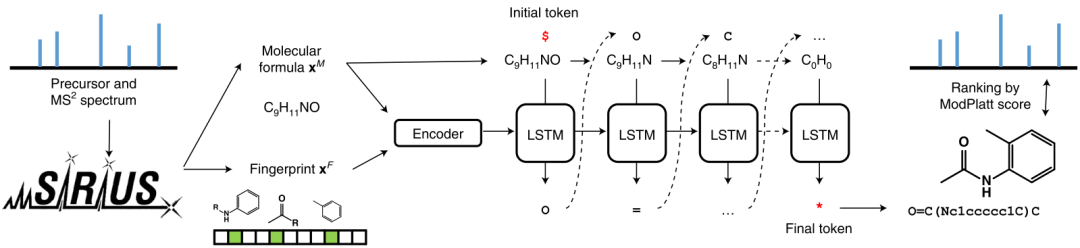
Figure 1: Framework of MSNovelist
4
Experiments
4.1 Evaluation Metrics
The evaluation metrics used in the experimental section of this article are as follows:
-
Valid SMILES Rate:
The ratio of generated SMILES samples that can be parsed by RDKIT;
-
Correct Match Rate:
The ratio of generated SMILES that can be parsed by RDKIT and match the molecular formula;
-
Modified Platt Score:
The modified Platt score calculated from the generated SMILES and the real mass spectrometry fingerprint, measuring the closeness of the generated candidates to the real fingerprint;
-
Similarity:
The Tanimoto similarity calculated between the predicted top-ranking candidate SMILES and the real molecular structure;
-
Precision:
The ratio of correct structures present in the predicted structures;
-
Top-n:
The ratio of correct structures in the top-n of the prediction results.
4.2 Comparative Experiments
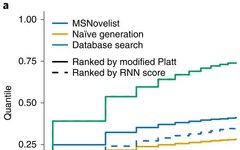
In order to verify the effectiveness of the MSNovelist model, a comparison was made with the database search algorithm CSI: FingerID on 3863 mass spectrometry data from the GNPS database, as shown in Figures 2 (a) and (c);
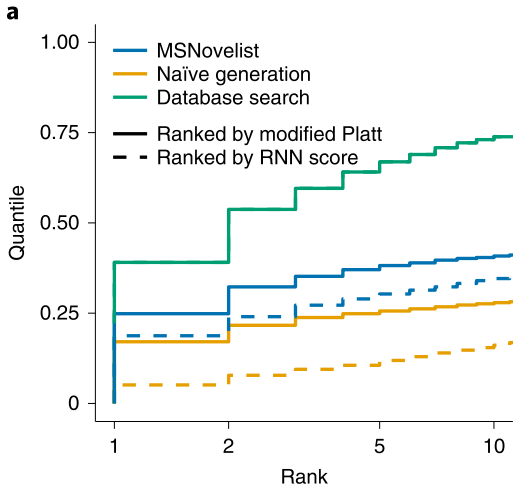
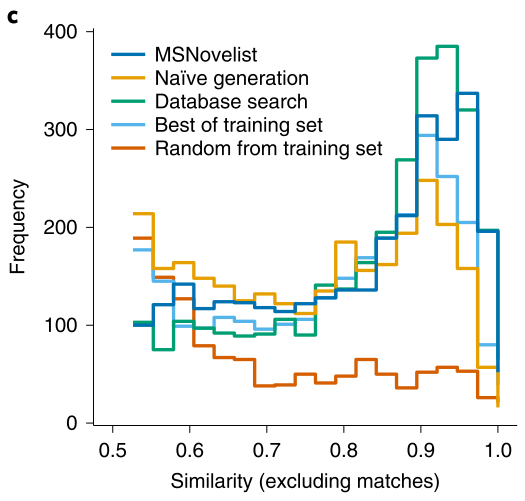
Figure 2: Comparison of MSNovelist model with other methods
From the comparison results, it can be seen that the MSNovelist model achieved better predictive performance than the database search algorithm CSI: FingerID.
4.3 Case Analysis
In order to verify the effectiveness of MSNovelist in predicting new molecular compound structures, it was applied to a dataset of 9 types of moss plants. Previous studies have shown that moss plants produce various secondary metabolites, but they have not been widely studied, providing potential opportunities for discovering natural products. This article extracted 576 tandem mass spectrometry from the MTBLS709 database, used the SIRIUS tool to infer their molecular formulas, and then predicted their molecular structures using the MSNovelist model, comparing it with the CSI: FingerID database search algorithm; the structures predicted by the MSNovelist model scored higher than the CSI: FingerID database search algorithm in 169 samples (75% of the samples), as shown in Figure 3 (a). Furthermore, one polyphenol compound (m/z 381.1020, molecular formula C21H16O7) was selected for visual analysis of the prediction results, as shown in Figure 4; from the visual results, it can be seen that compared to CSI: FingerID, MSNovelist can explain peaks 153 and 179, indicating that MSNovelist can more effectively predict new molecular compounds.
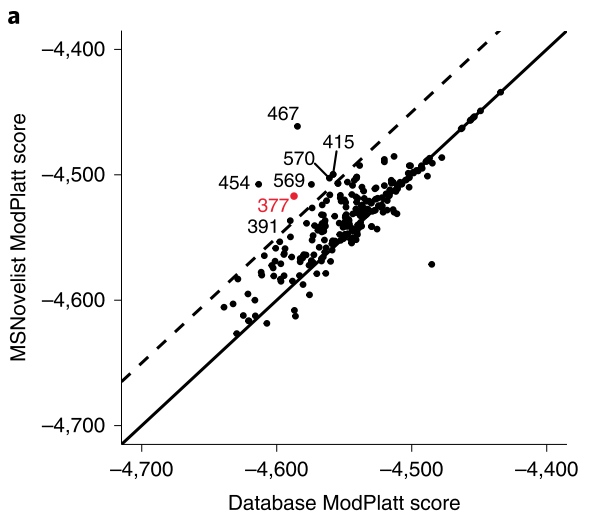
Figure 3: Comparison results on the moss plant dataset
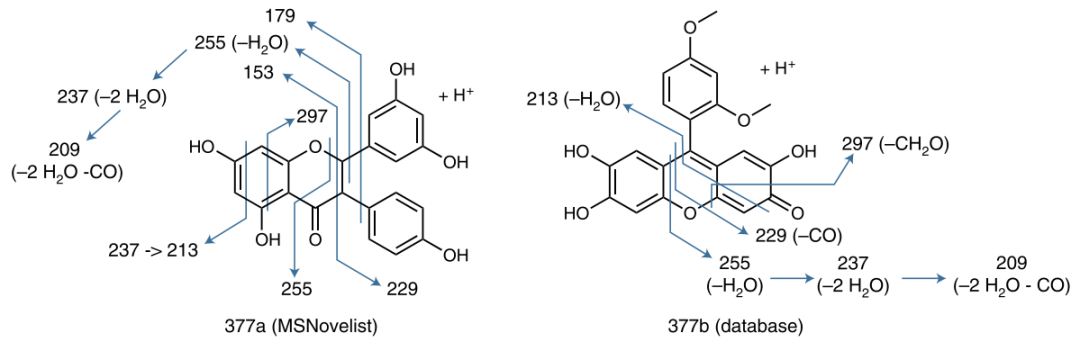
Figure 4: Visualization of prediction results for the polyphenol compound (m/z 381.1020, molecular formula C21H16O7)
5
Conclusion and Discussion
MSNovelist demonstrates that it is possible to generate molecular structures de novo from mass spectrometry without relying on structural databases. Although deep learning models have been used to generate candidate molecular structures from mass spectrometry data, MSNovelist is able to integrate encoded structural information into the fingerprints, proposing reasonable molecular structures for more than half of the MS2 mass spectrometry. Since MSNovelist relies on existing methods to determine molecular formulas, specifically the SIRIUS tool to predict the molecular formula of mass spectrometry, the error rate for determining molecular formulas for small molecular compounds with m/z < 300 is less than 10%. However, for molecular compounds with m/z up to 800, the error rate increases to over 50%, which can affect the predictive performance of the MSNovelist model.
References
Stravs, M.A., Dührkop, K., Böcker, S. et al. MSNovelist: de novo structure generation from mass spectra. Nat Methods (2022).
https://doi.org/10.1038/s41592-022-01486-3
Code
https://github.com/meowcat/MSNovelist.git
Disclaimer: The publication/reproduction of this article is solely for the purpose of disseminating information and does not imply endorsement of its content or verification of its authenticity. Any judgments made based on this content are at your own risk.If there is any infringement, please notify for deletion!
Long press to follow this public account

