
The field of new drug development has long been known for its lengthy timelines, high costs, significant risks, and low return on investment. The average development cost for a new drug has reached $2.6 billion, with an average time frame of ten years. Despite such high R&D costs and prolonged timelines, there is no guarantee that the developed drugs will successfully pass all clinical trials and reach the market. Even the development of generic drugs, which are generally easier, progresses very slowly.
With the continuous advancement of AI technologies such as deep learning, integrating AI with drug development can significantly reduce the time and costs associated with new drug development and accelerate the development and market entry of generic drugs. Undoubtedly, artificial intelligence and machine learning will usher in a faster, cheaper, and more efficient era of drug development.
Zilliz, in collaboration with leading global pharmaceutical research and development companies, has developed the MolSearch compound molecular structure analysis software, creating a new technological breakthrough for AI drug development.
| Milvus Vector Search Engine
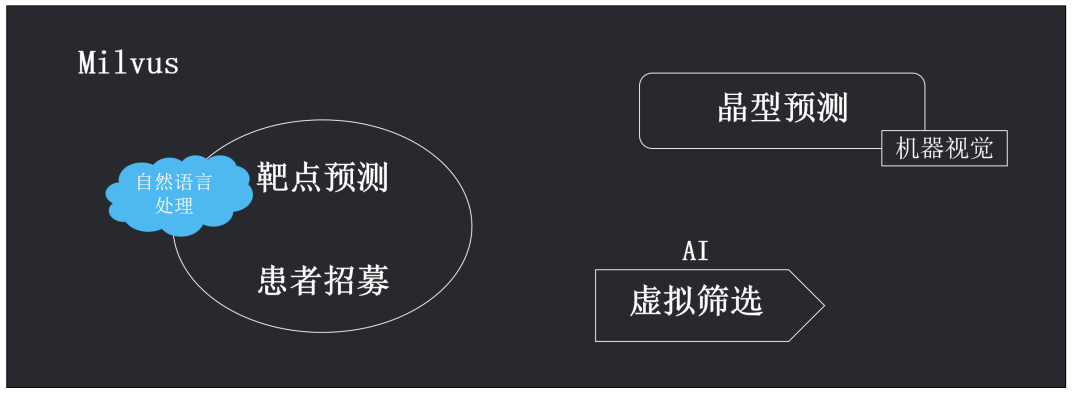
As an open-source feature vector similarity search engine, Milvus has been widely applied in key areas of AI technology, such as machine vision (image and video processing), natural language processing, and speech recognition, due to its powerful unstructured data processing capabilities.
| MolSearch Virtual Drug Screening Tool
MolSearch is an open-source compound analysis software developed based on the Milvus vector similarity retrieval engine, with the front-end design referencing the open-source software MolView[1]. For specific setup and functionality, please refer to https://github.com/zilliztech/MolSearch.
Drug chemistry experts typically optimize molecular modules based on scaffold jumps to design new drug structures for subsequent screening. Our initial intention in developing MolSearch was to accelerate virtual screening of massive compounds. Virtual screening is an essential and crucial step in new drug development, and its results significantly influence the success of later mouse experiments and clinical trials. The more extensive the compound database during virtual screening, the higher the accuracy of screening, thus increasing the likelihood of successful new drug development.
The molecular retrieval function in the MolSearch system, by integrating Milvus as the core retrieval engine, achieves the capability of performing second-level analysis on billions of chemical molecular structures, which is also a significant technological breakthrough for MolSearch in the field of drug development. Currently, MolSearch integrates an 820 million Zinc open chemical formula dataset[2], utilizing chemical formulas converted into 2048-bit chemical fingerprints (feature vectors) for high-performance vector calculations in Milvus to achieve similarity, substructure, and superstructure retrieval of molecular structures. The end-to-end retrieval performance is as follows:
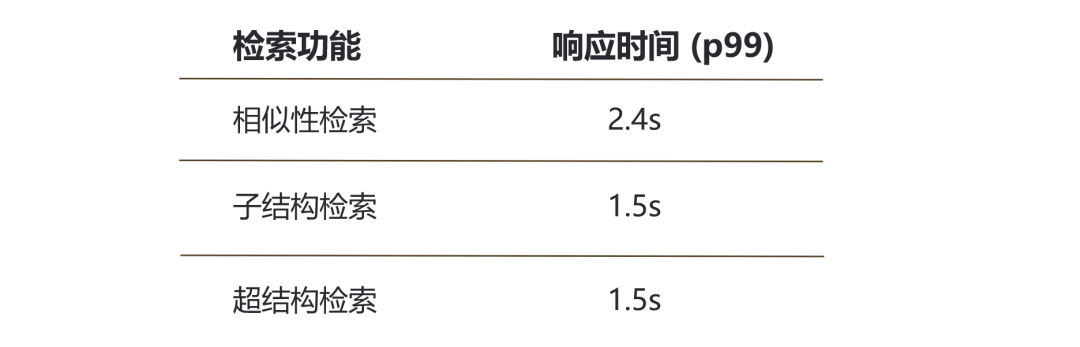
The p in the table represents the percentage, and the response time (p99) indicates how much time 99% of the retrieval can be completed.
| System Overview
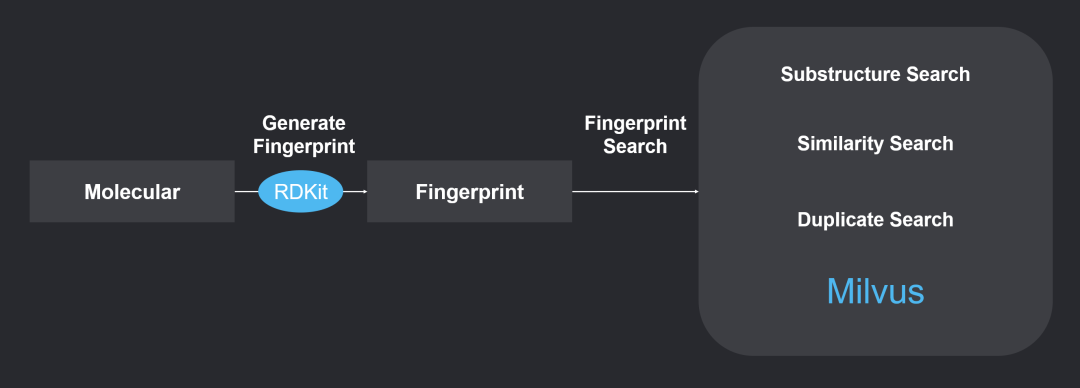
The virtual compound screening technology applied in the MolSearch system first converts the chemical formulas of compound molecules into chemical fingerprints (Chemical Fingerprint) using the RDKit tool[3], which is a set of feature vectors. Then, the distances between these vectors are calculated to analyze the similarities among compound molecules.
1. Chemical Fingerprint Generation
Chemical fingerprints are typically used for structure retrieval and similarity retrieval. As shown in the figure below, fingerprints are ordered lists represented in (1/0) bits, where each bit represents the presence of specified elements, molecular fragments, etc., in the chemical structure.
The MolSearch system utilizes the RDKit tool to generate RDKit fingerprints. The algorithm analyzes all molecular fragments along paths (usually linear) from one atom to a specified number of bonds, and hashes each path to produce a fingerprint. The image below displays all paths starting from NH2 (highlighted) with a length of 6, and each path is hashed into binary bits.
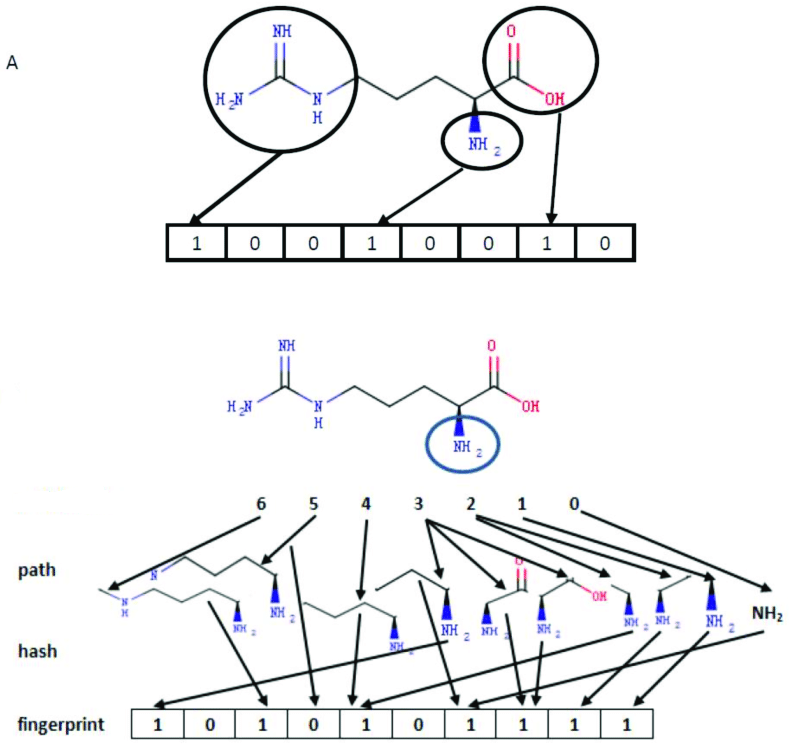
The above image only shows fragments and bits starting from a single initial atom. For a complete fingerprint, this process will be repeated for every atom in the molecule. Such fingerprints are applicable to any molecule and can specify fpSize to adjust the vector dimensions, and the final generated vectors can be imported into Milvus for retrieval.
from rdkit import Chem
mols = Chem.MolFromSmiles(smiles)
fp = Chem.RDKFingerprint(mols, fpSize=VECTOR_DIMENSION)
bits_fp = DataStructs.BitVectToFPSText(fp)
vectors = bytes.fromhex(hex_fp)2. Compound Retrieval
By importing the generated vectors into Milvus, a compound library can be established, enabling similarity retrieval, substructure retrieval, and superstructure retrieval based on different calculation methods.
from milvus import *
milvus = Milvus()
milvus.insert(collection_name=MILVUS_TABLE, records=vectors)
milvus.search(collection_name=MILVUS_TABLE, query_records=query_list, top_k=topk, params={})-
Similarity Retrieval
-
Substructure Retrieval
-
Superstructure Retrieval

Based on the above parameters, the calculation of chemical fingerprints can be described as follows: (for the complete formula, please click the CSDN link in the comments)

| Conclusion
| References
-
http://molview.org/
-
Sterling and Irwin, J. Chem. Inf. Model, 2015, https://pubs.acs.org/doi/abs/10.1021/acs.jcim.5b00559
-
Landrum, G. 2010. “RDKit.” Q2. https://www.rdkit.org/
