Hello everyone, I recommend a new article published in Molecular & Cellular Proteomics, with two corresponding authors, Jurgen Cox and Matthias Mann from the Max Planck Institute of Biochemistry. Professor Mann is a leading figure in the field of proteomics, and Jurgen Cox collaborated with him to develop all components of the MaxQuant ecosystem: the gold standard protein quantification software MaxQuant, the upstream database search engine Andromeda, and the statistical analysis software Perseus. In this article, the authors extend the MaxQuant ecosystem to the upstream of proteomics: mass spectrometry data acquisition. They developed a mass spectrometry data acquisition control software called MaxQuant.Live.
Although shotgun proteomics has become one of the most mature and efficient research methods in the field of proteomics, many issues and shortcomings still exist. Limited by the scanning speed of the mass spectrometer itself, only a portion of ions in the MS1 spectrum can be fragmented in the MS2 spectrum during experiments. Despite using data-dependent acquisition (DDA) methods to ensure experimental reproducibility, it still cannot guarantee that the same precursor ions will undergo MS2 acquisition in different parallel experiments. In contrast, using targeted fragmentation can ensure data acquisition reproducibility, but the number of ions that can be set in the list is limited. For many researchers, although Thermo’s official mass spectrometry control software Xcalibur is very user-friendly, it cannot flexibly set special acquisition methods for many specific experiments.
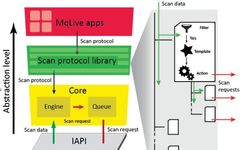
Therefore, in this article, the authors developed the MaxQuant.Live software, which can directly control any Thermo Fisher Q Exactive mass spectrometer through the instrument application programming interface (IAPI) developed by Thermo. Users can develop scan protocols through the MaxQuant.Live app and connect to the core program to directly control the data acquisition process. Users can freely design different logics to target different experimental purposes.
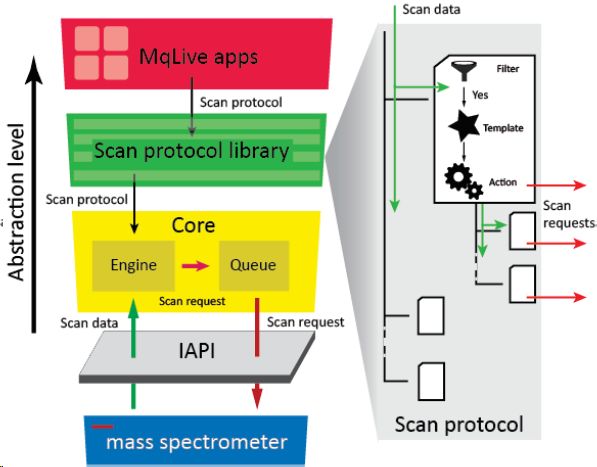
The authors first used the normal TOP15 logic to collect data with both MaxQuant.Live and Xcalibur, proving that the number of spectra (MS1 and MS2) collected by both methods is basically consistent, and the number of PSMs obtained from the database search is also roughly the same.
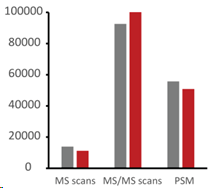
Then the authors conducted an interesting test using MaxQuant.Live, collecting data for 1000 peptides with 10 different NCEs, and comparing the fragmentation efficiency and conditions of different peptides under different NCEs in detail.
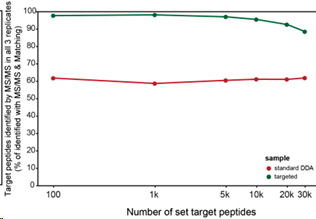
Finally, the authors also demonstrated that using MaxQuant.Live can effectively solve the problems mentioned earlier, allowing for targeted fragmentation of a large number of peptides simultaneously. The authors first conducted three normal DDA experiments and took the union of approximately 30,000 peptides to perform targeted fragmentation using MaxQuant.Live. To address the issue of having too many peptides that may need to be analyzed at the same time, the authors used dynamic normalization to reduce the retention time difference between the ions in the sample being analyzed and the reference sample, thereby minimizing the retention time window. The authors demonstrated that over 25,000 peptides can be stably fragmented in the three-targeted fragmentation scanning samples, achieving a parallelism that can rival DIA.
In summary, MaxQuant.Live has the potential to bring significant changes to the data acquisition modes of mass spectrometry in the future.
Article Author: GJJ
Article Link: https://doi.org/10.1074/mcp.TIR118.001131
Article Citation: DOI: 10.1074/mcp.TIR118.001131