
Click the blue text to follow us
Liver fibrosis is a common sequel of various chronic liver injuries, characterized by the excessive accumulation of extracellular matrix (ECM) proteins such as type I collagen and fibronectin. The persistence of the fibrotic process often leads to cirrhosis and hepatocellular carcinoma (HCC). The hallmark of liver fibrosis is the abnormal activation and proliferation of hepatic stellate cells (HSCs), which can be mediated by various extracellular signals, including hepatocytes, liver sinusoidal endothelial cells (LSECs), natural killer (NK) cells, and macrophages. Currently, there are limited antifibrotic therapies specifically targeting HSC activation. Therefore, expanding research on the fibrotic process of chronic liver disease is crucial for developing effective therapies against liver fibrosis.
Macrophages are considered key drivers of fibrosis in different organs (including the liver). Numerous studies have shown that liver macrophages are closely related to the transdifferentiation of HSCs into collagen-producing myofibroblasts. Liver macrophages can be divided into resident macrophages (Kupffer cells) and recruited monocyte-derived macrophages, both of which play critical roles in HSC activation. In the early stages of liver fibrosis, liver macrophages are activated and release various pro-inflammatory and pro-fibrotic factors, such as TNF-α, IL-1β, chemokine CCL2, TGF-β, and platelet-derived growth factor (PDGF), to regulate HSC activation.
Members of the FGF protein family can be classified into secreted (FGF1-10 and FGF15-23) and intracellular non-secreted types (FGF11-14). Among them, FGF12 is highly expressed in the nervous system, regulates voltage-gated sodium channels in neurons, and influences the NF-κB signaling pathway and MAPK signaling pathway by binding to specific proteins. Recent studies have also found that FGF12 is an important regulatory factor in vascular smooth muscle cell homeostasis, thus affecting the progression of atherosclerosis and pulmonary hypertension. However, research on the role of FGF12 in liver homeostasis is limited, and whether FGF12 can regulate liver fibrosis is largely unknown.
Academician Li Xiaokun’s team from Wenzhou Medical University published a research paper titled “Macrophage-specific FGF12 promotes liver fibrosis progression in mice” in Hepatology in March 2023, revealing that macrophage-specific FGF12 activates HSCs through the MCP-1/CCR2 axis, playing a pro-fibrotic role in a mouse model.
“
FGF12 is upregulated in mouse liver fibrosis models induced by BDL and CCL4
To investigate whether FGF12 is associated with liver fibrosis, the authors first detected the expression of FGF12 in two mouse liver fibrosis models induced by BDL and chronic CCL4 injection. In the liver tissues of mice subjected to BDL surgery and CCL4 treatment, the mRNA levels of the HSC activation marker Acta2 and FGF12 were significantly upregulated (Figure 1A, B). The WB results were consistent, showing that FGF12 protein expression was upregulated in both liver fibrosis models. Additionally, FGF12 immunohistochemistry (IHC) staining showed increased expression of FGF12 in the livers of BDL and CCL4-induced fibrosis models (Figure 1C, D). From the immunohistochemical images of fibrotic livers, it can be observed that FGF12 expression accumulates in fibrotic liver areas and non-parenchymal liver cells (Figure 1C, D). These data indicate that FGF12 is upregulated in fibrotic livers, increasing the likelihood of its role in the progression of liver fibrosis.
To clarify the molecular mechanism by which FGF12 exerts its effects in liver fibrosis, the authors first studied the cellular distribution of FGF12. They isolated hepatocytes and non-parenchymal liver cells from the livers of BDL-induced and CCL4-induced fibrotic mice, and WB results confirmed that FGF12 is mainly expressed in non-parenchymal liver cells, not in hepatocytes (Figure 1E, F). Then, in the livers of BDL-treated mice, immunofluorescence co-localization staining was performed using α-SMA, F4/80 (macrophage marker), or CD31 (LSEC marker) with FGF12. The results indicated that FGF12 upregulation mainly occurs in macrophages compared to HSCs and LSECs in fibrotic liver tissues (Figure S1A). Similarly, in the livers of mice with CCL4-induced fibrosis, the results were consistent (Figure S1B). In summary, these data suggest that liver macrophages are the primary contributors to FGF12 expression during the process of liver fibrosis in mice.
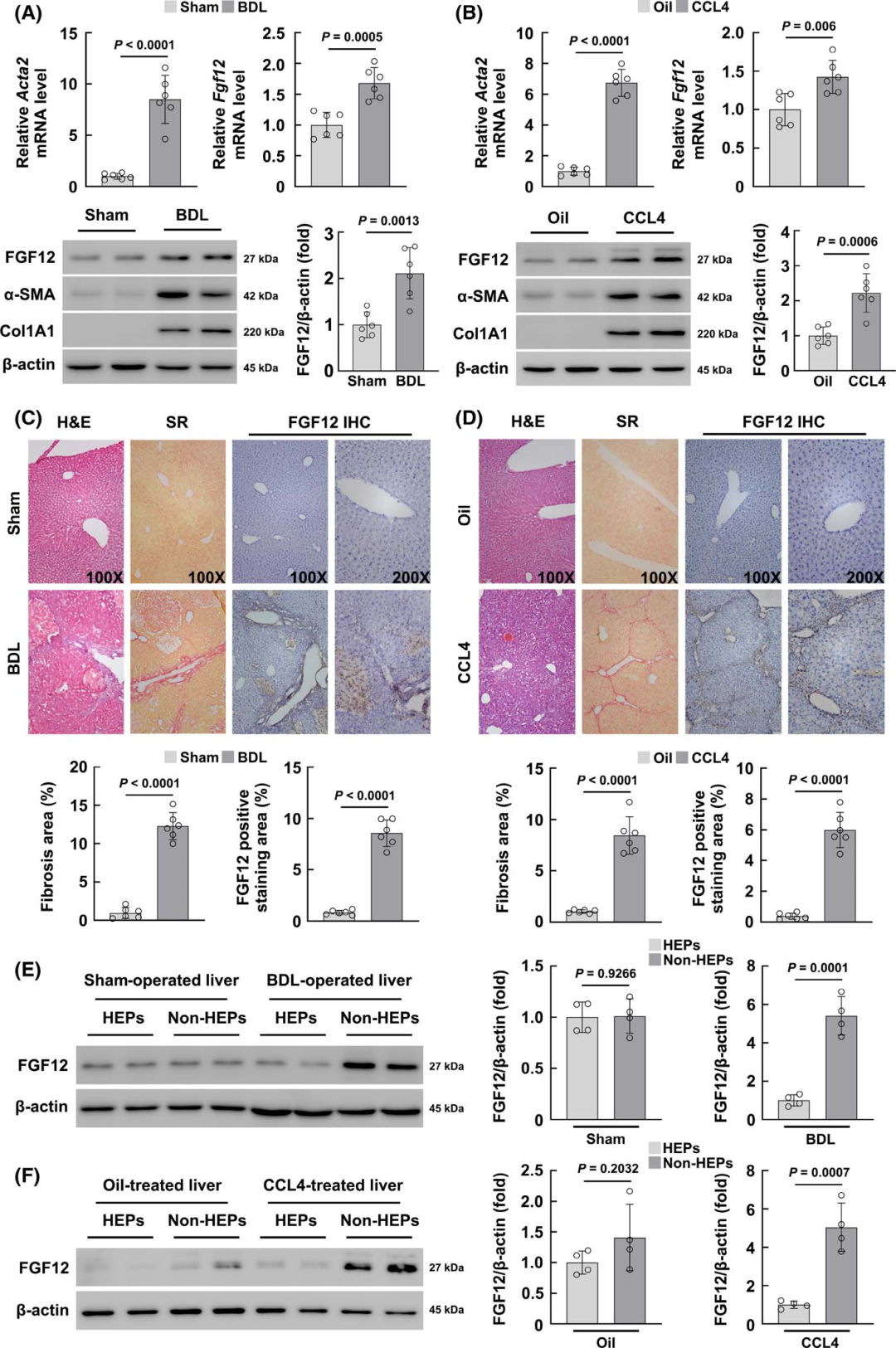
Figure 1 FGF12 is upregulated in mouse liver fibrosis models
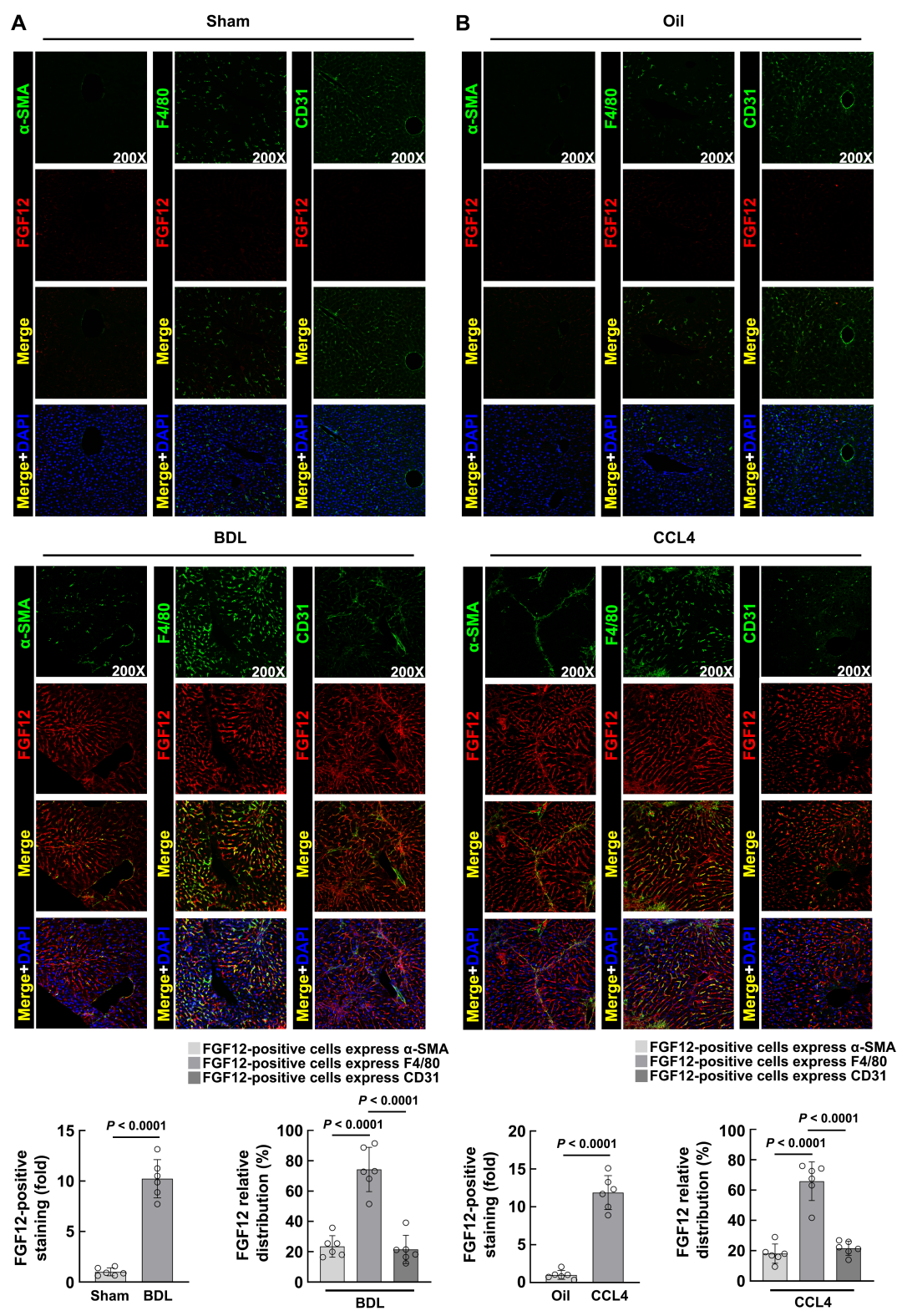
Figure S1 FGF12 is mainly expressed in macrophages in mouse fibrotic liver
“
Macrophage FGF12 knockout alleviates BDL and CCL4-induced liver fibrosis in mice
To study the role of FGF12 in liver fibrosis, the authors constructed myeloid-specific FGF12 knockout mice (Fgf12 fl/fl;Lyz2cre+/−) to knock out FGF12 in macrophages. The littermate Fgf12 fl/fl mice were used as controls. The authors then subjected these mice to BDL, and Sirius Red staining showed a significant reduction in fibrotic areas in Fgf12 fl/fl;Lyz2cre+/− mice (Figure 2A), and serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were also significantly reduced (Figure 2B). Hydroxyproline assay results also indicated reduced collagen deposition in the livers of BDL-treated Fgf12 fl/fl;Lyz2cre+/− mice (Figure 2C). These results indicate that compared to control mice, liver fibrosis is attenuated in Fgf12 fl/fl;Lyz2cre+/− mice after BDL, suggesting that FGF12 may promote the development of liver fibrosis in mice.
To confirm the results, the authors performed α-SMA and type I collagen immunofluorescence staining to assess the degree of fibrosis. The results showed that the absence of FGF12 in macrophages reduced the fibrotic area after BDL, while there was no significant change in normal liver (Figure 2D). WB results also indicated that pro-inflammatory factor TNF-α and pro-fibrotic factor TGF-β levels were lower in BDL-treated Fgf12 fl/fl;Lyz2cre+/− mice compared to Fgf12 fl/fl mice, while α-SMA and Col1A1 were reduced (Figure 2E). Subsequently, the authors also induced liver fibrosis in mice with CCL4, and the results were consistent with those observed in the BDL mouse model, showing that macrophage FGF12 knockout significantly improved liver fibrosis in mice (Figures 3A-E). In summary, these data reveal that macrophage FGF12 plays a negative role in the process of liver fibrosis in mice.
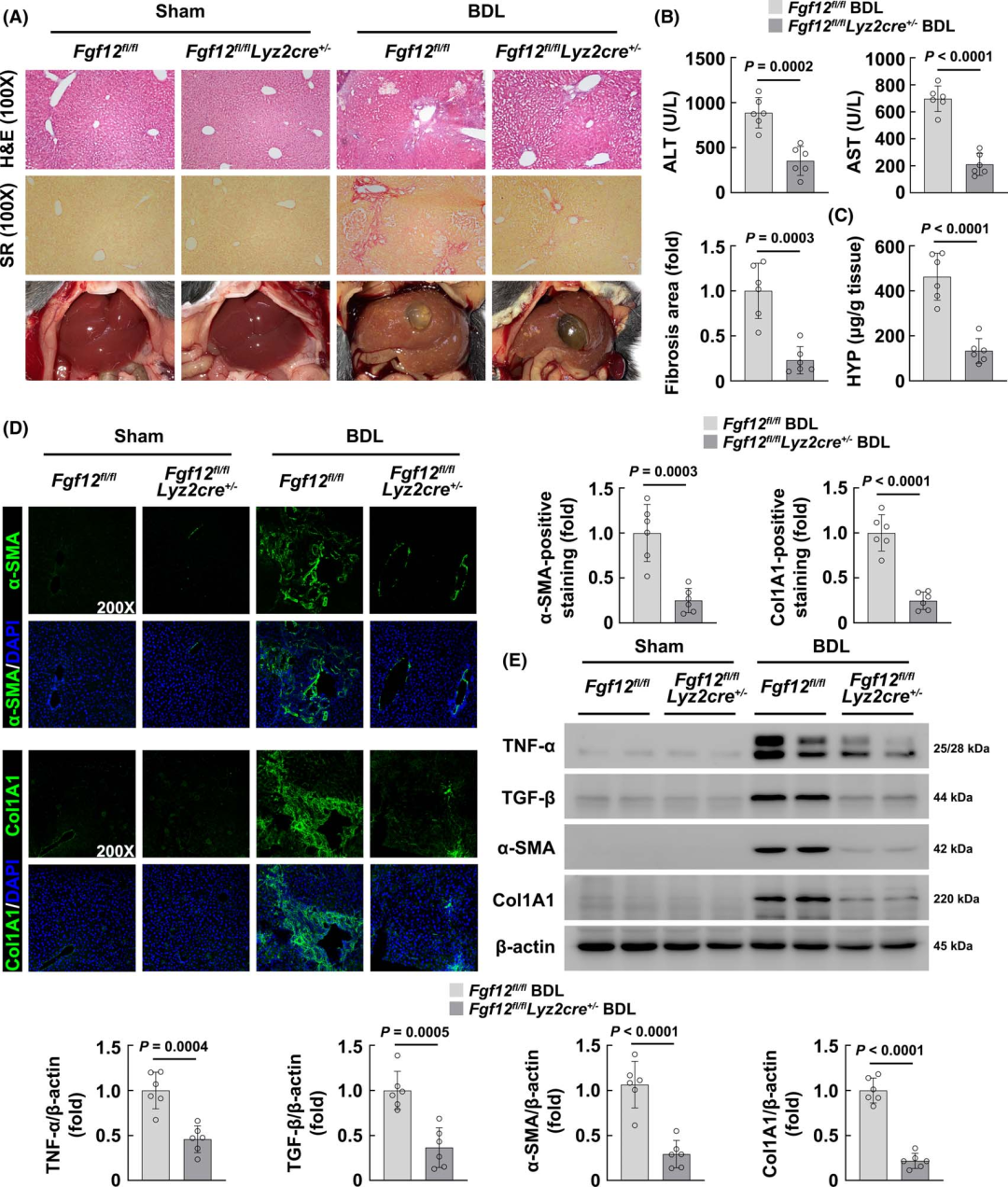
Figure 2 Macrophage FGF12 knockout alleviates BDL-induced liver fibrosis in mice
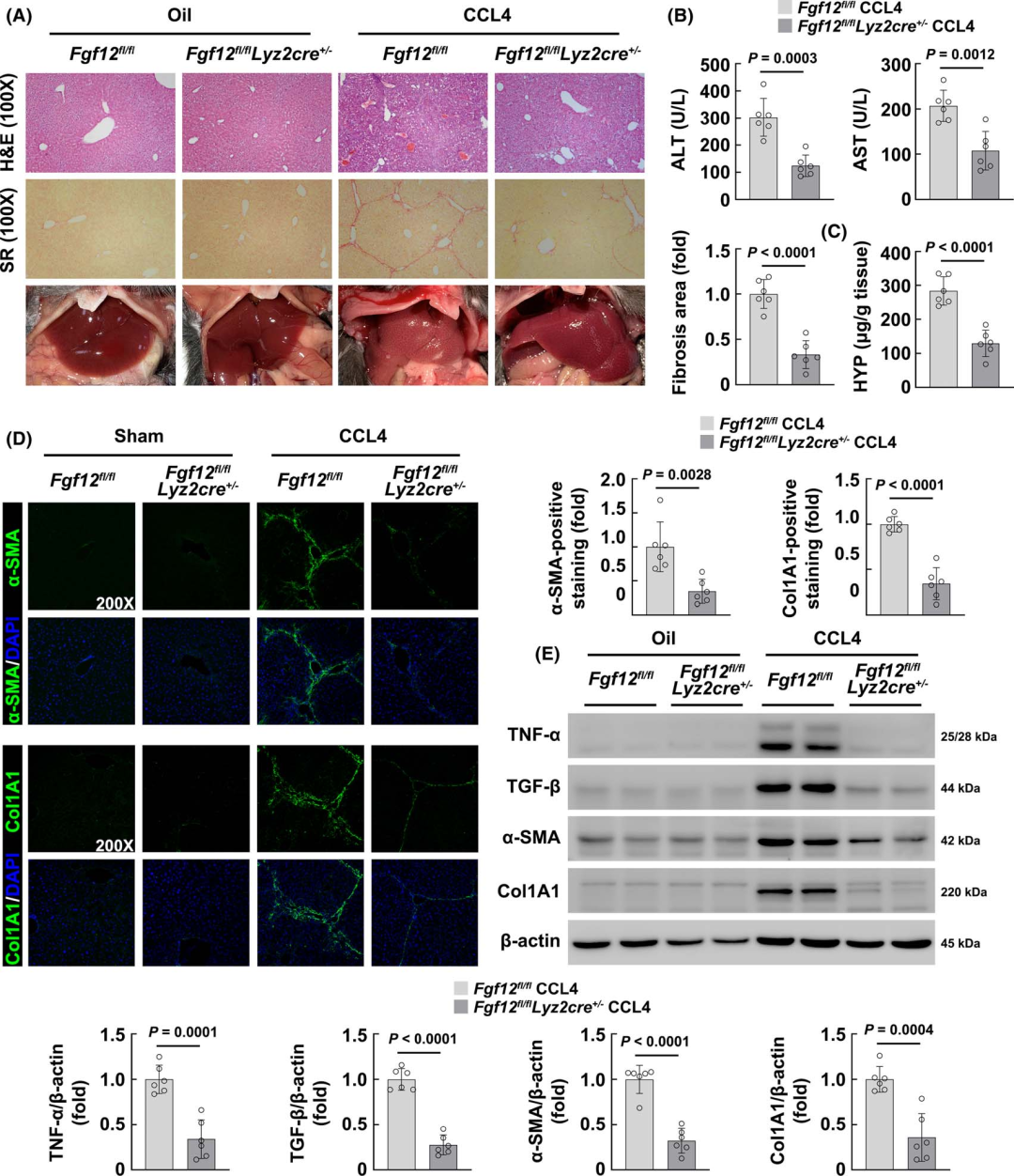
Figure 3 Macrophage FGF12 knockout alleviates CCL4-induced liver fibrosis in mice
“
FGF12 regulates macrophage phenotype and cytokine and chemokine responses in mouse liver fibrosis
As macrophage FGF12 has the ability to regulate the liver fibrosis process, the authors first hypothesized that the expression of FGF12 may influence the phenotypic transition of macrophages during liver fibrosis. Considering that bone marrow-derived Ly6C high expressing macrophages dominate the macrophage population in injured livers and are crucial for HSC activation, the authors first examined this phenotypic change in mouse models. Flow cytometry analysis showed that BDL surgery and CCL4 treatment significantly upregulated the number of Ly6C high expressing macrophages, while in the livers of Fgf12 fl/fl;Lyz2cre+/− mice, the number of Ly6C high expressing macrophages significantly decreased after BDL or chronic CCL4 injection (Figures 4A, B). Meanwhile, the absence of FGF12 did not change the number of Ly6C cell populations, indicating that the absence of FGF12 induced a phenotypic transition of macrophages from Ly6C high expressing to Ly6C low expressing during liver fibrosis in mice.
During liver fibrosis, in addition to infiltrating monocyte-derived macrophages, resident liver macrophages (Kupffer cells) also play an important role in the development of fibrosis. However, the data show that LPS/IFN-γ can more significantly induce FGF12 upregulation in bone marrow-derived macrophages (Figure S2), indicating that FGF12 may play a more important role in regulating the function of monocyte-derived macrophages.
To further understand the role of FGF12 in the regulation of liver fibrosis, the authors performed RNA sequencing analysis on Fgf12 fl/fl;Lyz2cre+/− mice and control Fgf12fl/fl mice after BDL . KEGG pathway enrichment analysis indicated that the downregulated genes mediated by FGF12 deficiency were mainly involved in cytokine and chemokine responses, as well as ECM receptor interactions, which is also the most significantly upregulated pathway after BDL surgery (Figures 4C, D). As shown in the heat map results, these representative cytokine and chemokine-related genes and ECM-related differentially expressed genes were significantly downregulated in BDL-treated Fgf12 fl/fl;Lyz2cre+/− mice (Figure S3), indicating that the inflammatory and fibrotic responses were reduced in macrophage FGF12 knockout mice.
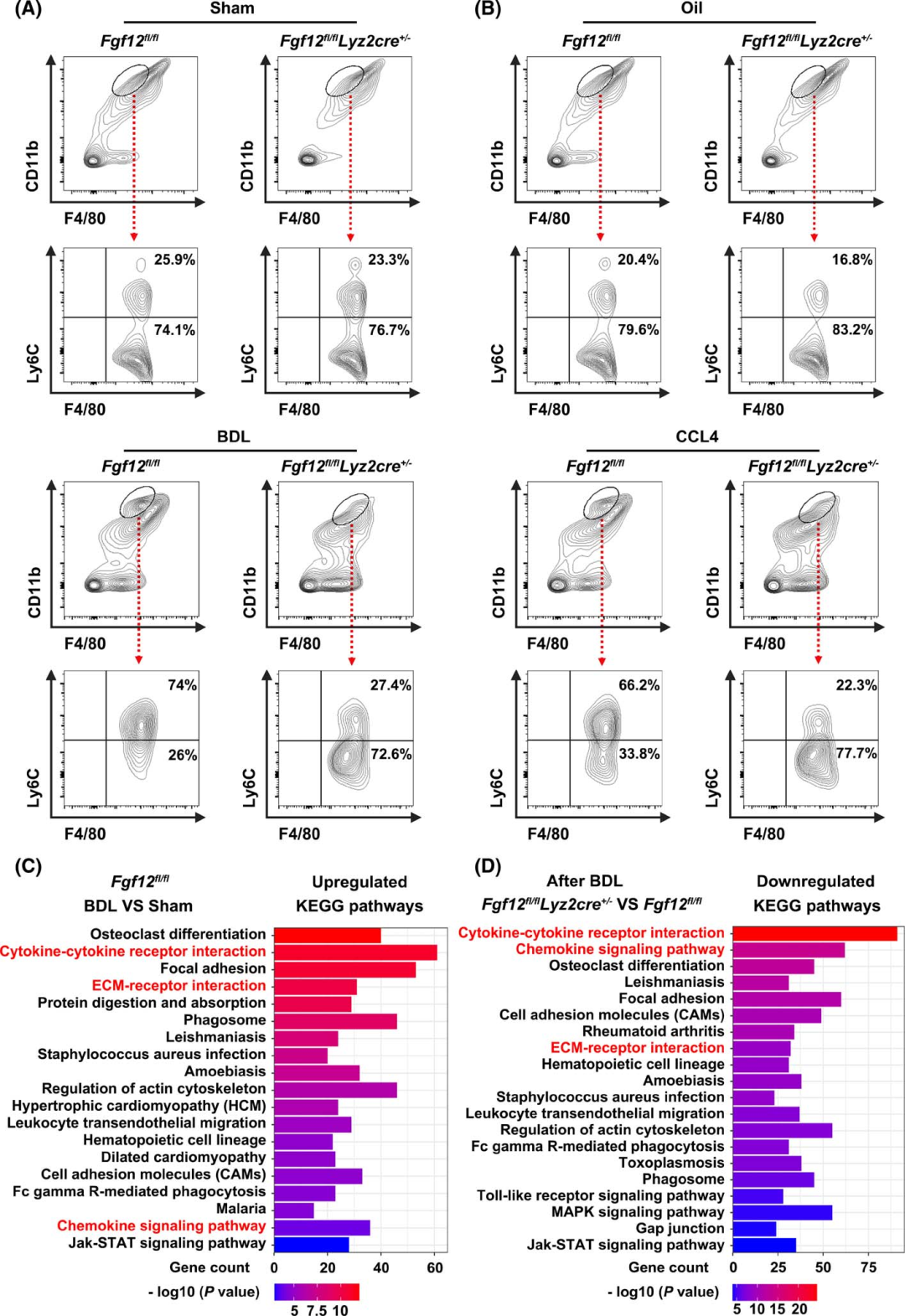
Figure 4 FGF12 regulates macrophage phenotype and cytokine and chemokine responses
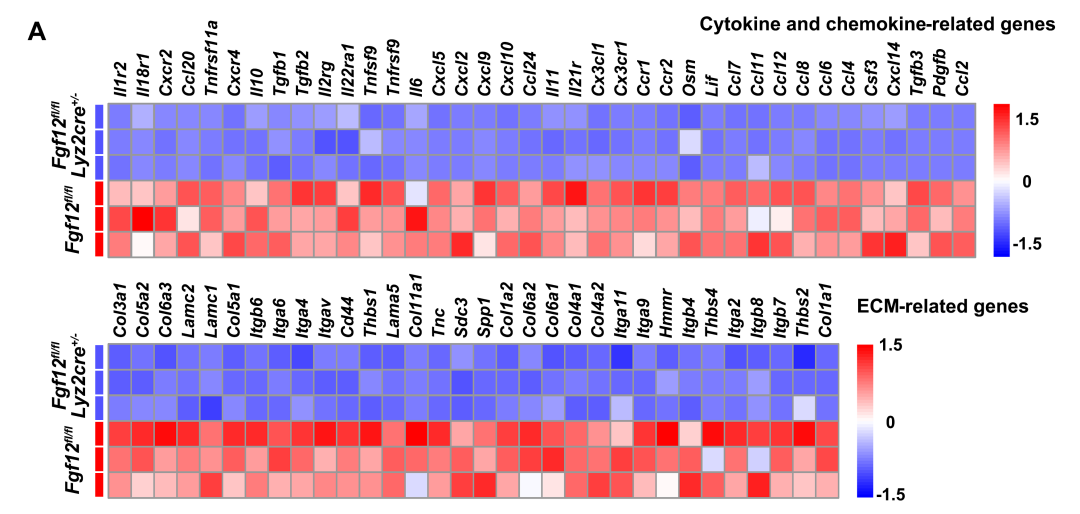
Figures S2-S3
“
The expression of macrophage-derived MCP-1 plays an important role in FGF12-mediated exacerbation of fibrosis
Next, the authors attempted to explore how macrophage FGF12 regulates HSC activation. To investigate the mechanisms involved in this process, the authors first searched for the most significantly changed genes in the liver tissues of Fgf12 fl/fl;Lyz2cre+/− mice and Fgf12fl/fl mice after BDL. Volcano plot gene expression analysis indicated that Ccl2 is one of the most significantly downregulated genes (Figure 5A). WB analysis also consistently showed that after BDL or chronic CCL4 injection, the protein expression of MCP-1 (Ccl2) was significantly reduced in Fgf12 fl/fl;Lyz2cre+/− mice (Figure 5B).
In in vitro studies, the authors isolated BMDMs and treated them with LPS/IFN-γ to induce pro-inflammatory activation. They then used lentivirus to knock out and overexpress FGF12 in BMDMs, finding that the knockdown or overexpression of FGF12 did not affect the morphology and activation of normal macrophages (iNOS immunofluorescence) (Figures 5C, D). However, knockdown of FGF12 significantly alleviated LPS/IFN-γ-induced macrophage activation (Figure 5C), while overexpression of FGF12 exacerbated activation (Figure 5D). Furthermore, the authors measured the MCP-1 levels in the culture supernatant of macrophages, and the results showed that after LPS/IFN-γ treatment, FGF12 knockdown reduced the secretion of MCP-1, while overexpression increased the secretion of MCP-1 from activated macrophages (Figures 5C, D). Most importantly, flow cytometry analysis showed that in LPS/IFN-γ-treated BMDMs, FGF12 knockdown induced the transition from Ly6C high expressing phenotype to Ly6C low expressing phenotype (Figure 5E), while FGF12 overexpression promoted the transition of activated macrophages to Ly6C high expressing phenotype (Figure 5F). Overall, these data suggest that macrophages with FGF12 regulation play an important role in the progression of liver fibrosis in mice by modulating MCP-1 expression.
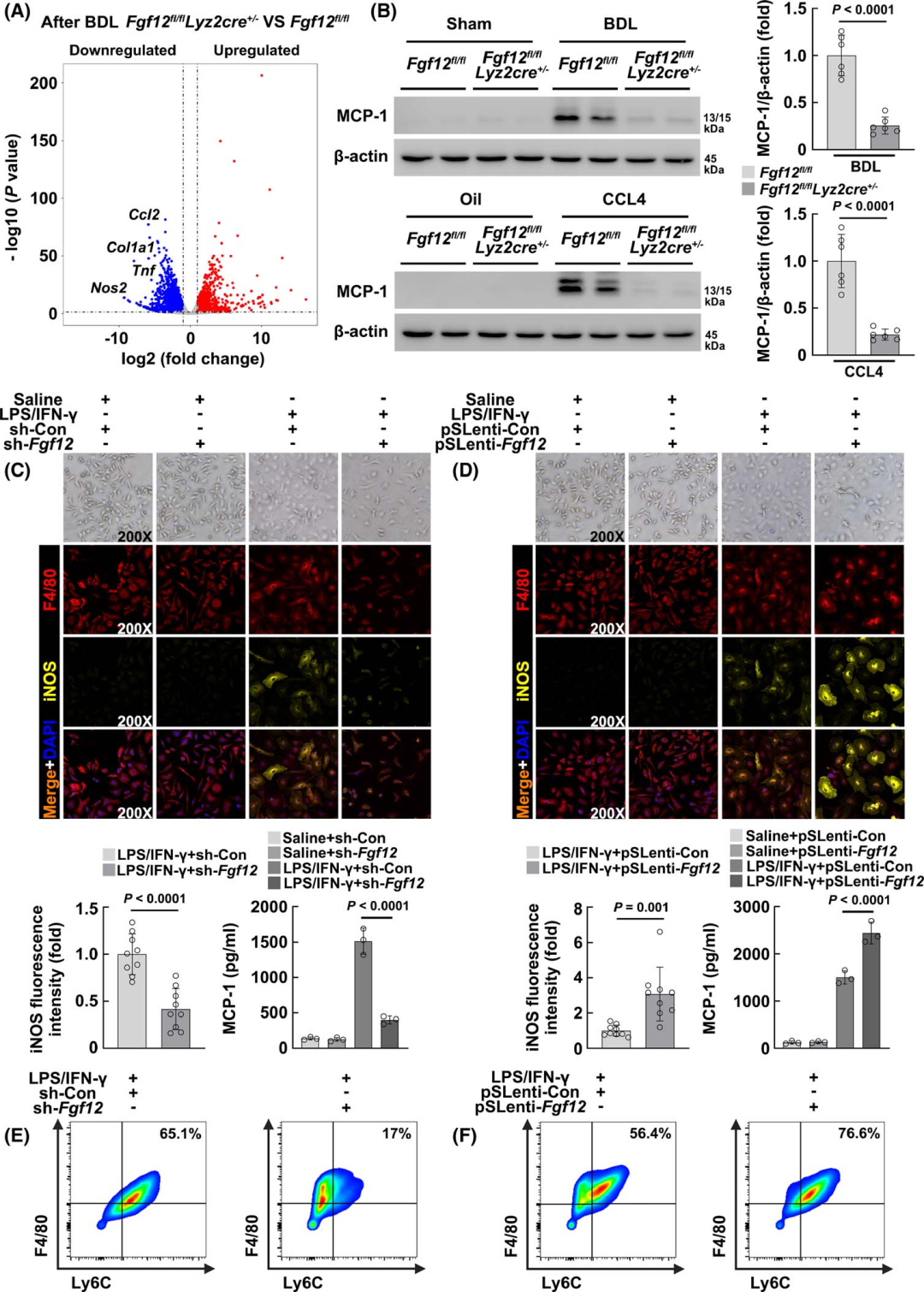
Figure 5 The expression of macrophage-derived MCP-1 plays an important role in FGF12-mediated exacerbation of fibrosis
“
Macrophage FGF12 regulates HSC activation through the MCP-1/CCR2 axis
To further confirm that FGF12 regulates the expression of MCP-1 in macrophages, thereby affecting HSC activation, the authors conducted in vitro co-culture experiments. They first isolated primary HSCs from mice and stimulated them with conditioned media (CM) collected from cultured BMDMs (Figure 6A). Considering that in in vitro studies, simply using CM from pro-inflammatory activated macrophages to culture HSCs is difficult to induce their activation, they treated HSCs with a low dose of recombinant mouse TGF-β to simulate in vivo fibrotic conditions. Compared to the control group, CM from LPS/IFN-γ-treated BMDMs promoted HSC activation, evident in morphological changes and upregulation of α-SMA fluorescence intensity and Acta2 (α-SMA) mRNA levels (Figure 6B). Furthermore, knockdown of FGF12 inhibited the ability of macrophages to promote HSC activation, while treatment with recombinant mouse MCP-1 (rMCP-1) again promoted activation (Figure 6B). Additionally, WB analysis detected the protein expression levels of α-SMA and type I collagen, as well as the activation of the MEK/Erk signaling pathway, which is known to be a downstream signal activated by MCP-1. The results showed that CM from LPS/IFN-γ-treated BMDMs upregulated α-SMA and type I collagen expression in HSCs, while knockdown of FGF12 in BMDMs significantly inhibited this phenomenon, and treatment with rMCP-1 reactivated HSCs, with similar changes observed in phosphorylated MEK1/2 and Erk1/2 (Figure 6C). Compared to the control group, CM collected from BMDMs overexpressing FGF12 further promoted HSC activation, as evidenced by increased α-SMA fluorescence intensity, Acta2 mRNA levels, and α-SMA/type I collagen expression (Figures 6D, E).
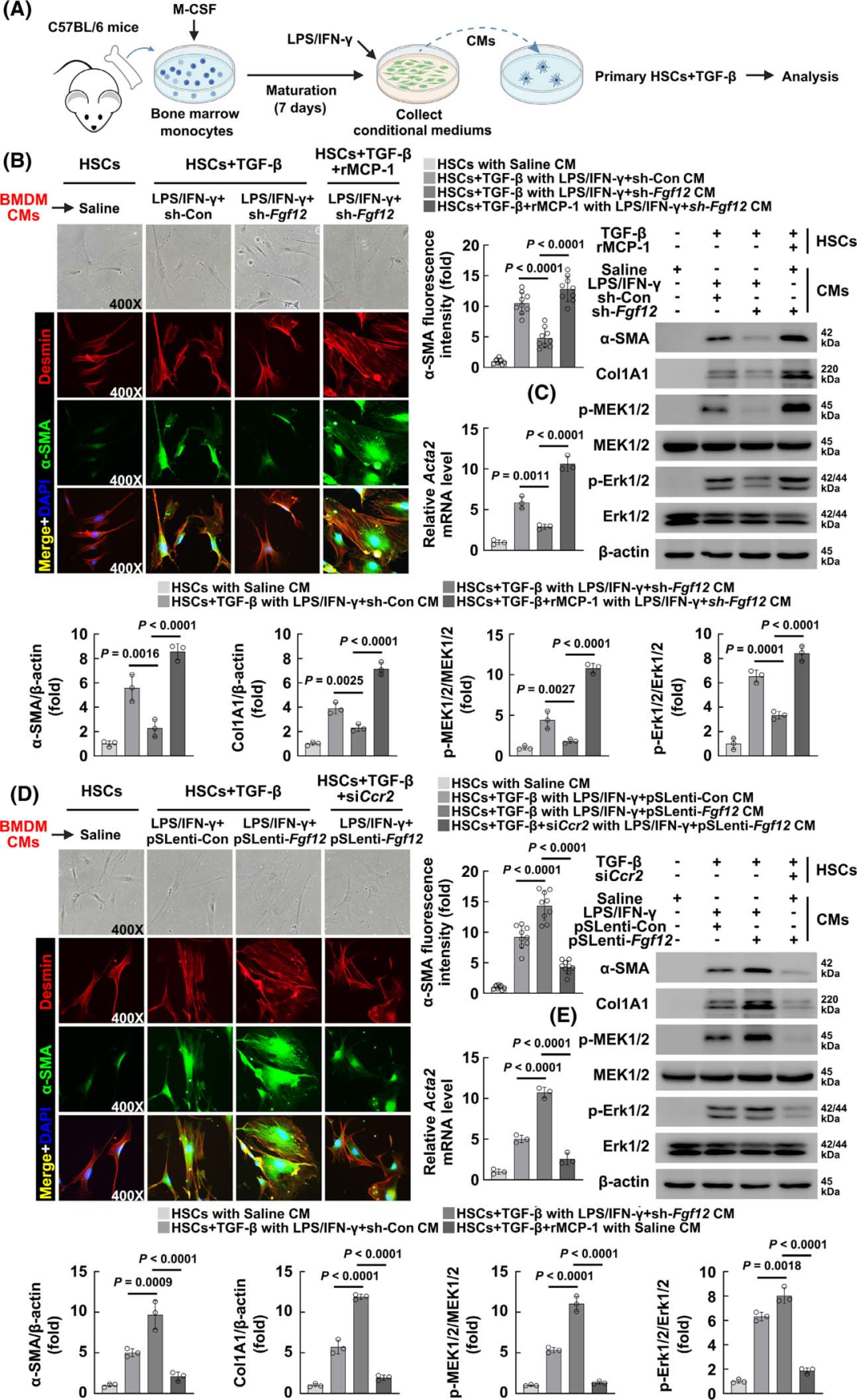
Figure 6 Macrophage FGF12 regulates HSC activation through the MCP-1/CCR2 axis
“
FGF12 regulates pro-inflammatory activation of macrophages mainly through the Jak-STAT pathway
Next, the authors attempted to elucidate the molecular mechanisms by which FGF12 regulates pro-inflammatory activation of macrophages. RNA sequencing analysis was performed on FGF12 gene knockout BMDMs treated with LPS/IFN-γ, and KEGG pathway enrichment analysis indicated that in control BMDMs, LPS/IFN-γ treatment significantly induced the upregulation of cytokine and chemokine responses, as well as Jak-STAT signaling activation (Figure 7A), while in FGF12-silenced macrophages, in addition to significantly inhibiting cytokine and chemokine responses, Jak-STAT signaling was also significantly downregulated (Figure 7B). Subsequently, the authors confirmed the RNA sequencing results using WB, showing that the loss and gain of function of FGF12 did not affect the phosphorylation of STAT1 and STAT3 in control BMDMs (Figures 7C, D). However, the activation of STAT1/STAT3 after LPS/IFN-γ treatment was significantly inhibited by FGF12 knockout, while overexpression of FGF12 increased activation, also having a similar effect on iNOS protein expression (Figures 7C, D), indicating that FGF12 regulates STAT activation under pathological conditions, affecting pro-inflammatory activation of macrophages.
To confirm these results, the authors treated LPS/IFN-γ-stimulated FGF12-overexpressing BMDMs with the Jak-STAT signaling inhibitor INCB18424, and the results showed that inhibiting Jak-STAT signaling significantly reduced the expression of iNOS and MCP-1 in FGF12-overexpressing macrophages (Figure 7E). Most importantly, INCB18424 treatment also promoted the transition of FGF12-overexpressing macrophages to the Ly6C low expressing phenotype (Figure 7F). These results confirm that the pro-inflammatory activation regulated by FGF12 in macrophages is mainly mediated through Jak-STAT signaling activation.
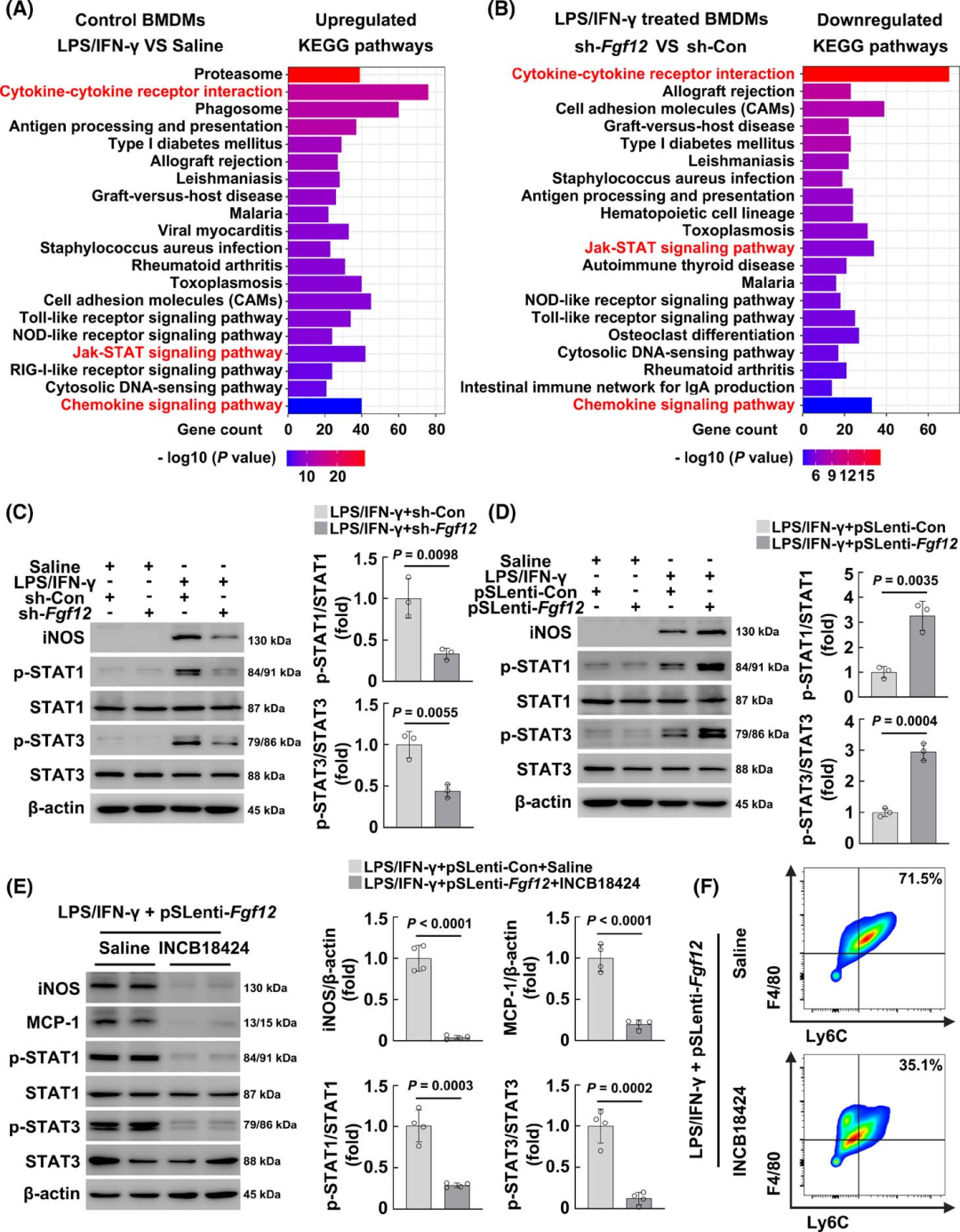
Figure 7 FGF12 regulates pro-inflammatory activation of macrophages mainly through the Jak-STAT pathway
“
FGF12 expression increases in human fibrotic liver tissues
To verify the potential clinical relevance of the above findings, the authors examined the expression levels of FGF12 in human liver samples. Through immunofluorescence staining, the authors found that FGF12 expression positively correlated with the stage of liver fibrosis (Ishak scoring) and the expression of α-SMA (Figures 8A-C). Additionally, FGF12 mRNA expression also positively correlated with ACTA2 expression (Figure 8D), suggesting that FGF12 may play a role in human liver fibrosis. The authors then performed immunofluorescence co-localization staining of FGF12 with F4/80, and the results showed that FGF12 was expressed in liver macrophages in human cirrhotic livers (Figure 8E). In summary, these data indicate that the expression of FGF12 in liver macrophages is upregulated in fibrotic human liver tissues and suggest that the upregulation of FGF12 may play a role in the progression of human liver fibrosis.
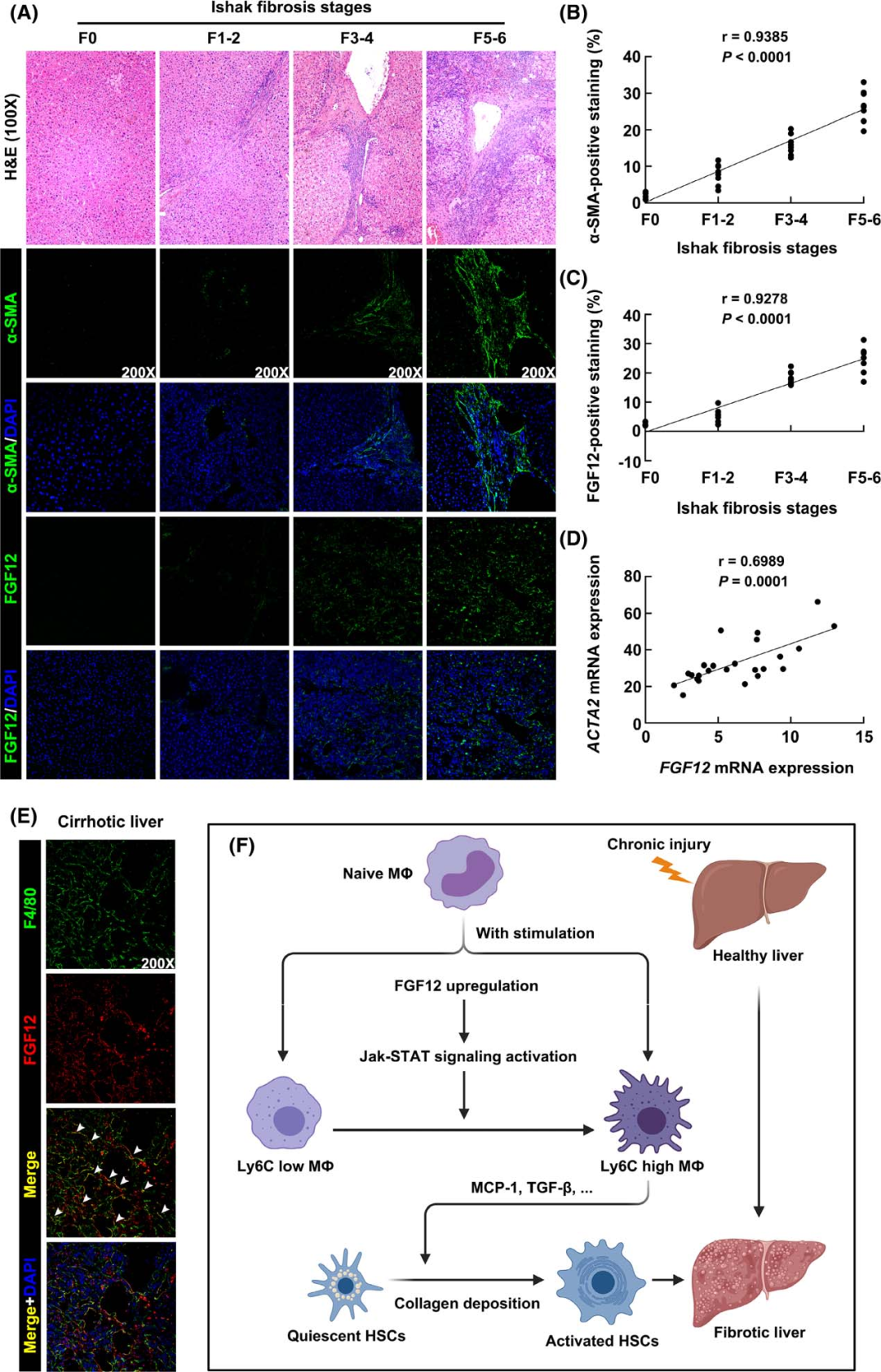
Figure 8 FGF12 expression increases in fibrotic human liver tissues
This study used two mouse liver fibrosis models, bile duct ligation (BDL) and chronic CCL4 injection, to investigate the pro-fibrotic role of macrophage FGF12 in the progression of liver fibrosis. The results indicated that FGF12 knockout reduced the number of Ly6C high expressing macrophages in fibrotic liver tissues of mice, thereby inhibiting HSC activation. Mechanistically, the authors found that FGF12 could regulate pro-inflammatory activation of macrophages through the Jak-STAT pathway. Furthermore, by analyzing human clinical liver samples, they confirmed that FGF12 is upregulated in human liver fibrosis and correlated with disease severity.
In summary, this study suggests that macrophage FGF12 knockout can inhibit HSC activation and the progression of liver fibrosis. FGF12 may be an important factor in regulating macrophage activation, and targeting macrophage FGF12 represents a potential therapeutic strategy against liver fibrosis.
Authors of this article
Qian Jialong
China Pharmaceutical University, Master of Pharmacology

References:
Li S, Zhou B, Xue M, et al. Macrophage-specific FGF12 promotes liver fibrosis progression in mice. Hepatology. 2023;77(3):816-833.