
————————————
Exclusive arrangement, unauthorized use will be pursued
In life science research, the interaction between proteins and nucleic acids plays a crucial role. Accurately predicting nucleic acid binding sites on protein surfaces is significant for understanding life regulatory mechanisms and developing new drugs. Recently, a research team from the University of Hong Kong and ShanghaiTech University proposed an innovative deep learning method—NesT-NABind (Nested Transformer for Nucleic Acid-Binding site prediction), which has achieved significant breakthroughs in the task of protein-nucleic acid binding site prediction.
Research Background and Significance
The binding of proteins to nucleic acids is decisive for key biological processes such as gene regulation, transcription, and translation. Traditional experimental methods (such as X-ray crystallography, nuclear magnetic resonance, and cryo-electron microscopy) can provide atomic-level structural information, but these methods are often time-consuming and labor-intensive, with technical limitations. Therefore, developing efficient and accurate computational methods to predict protein-nucleic acid binding sites is of great practical significance.
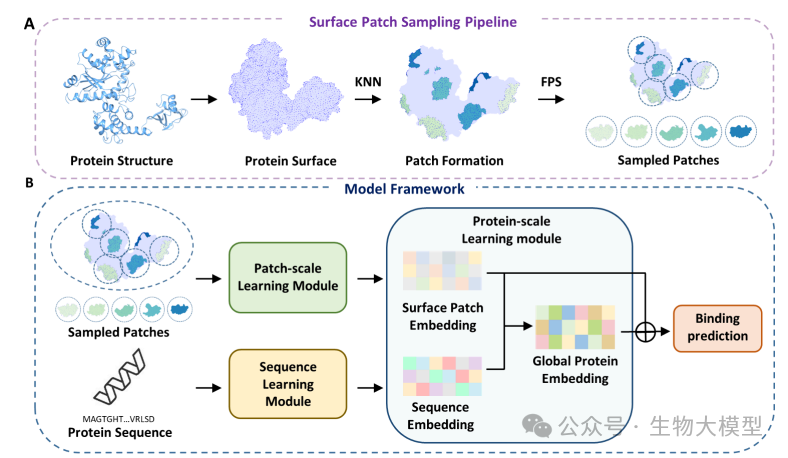
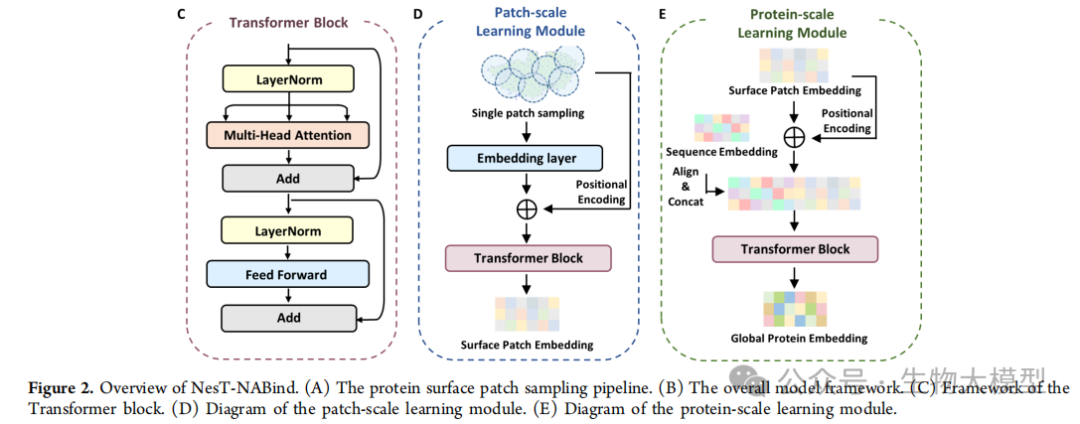
Figure 2 of the paper shows the overall architecture of NesT-NABind, including the surface fragment sampling process, model framework, and detailed structure of each module. The model innovatively adopts a nested Transformer structure, processing protein information on both local and global scales.
Technical Innovation and Method
1. Multi-Scale Feature Extraction
The core innovation of NesT-NABind lies in its unique nested Transformer architecture, specifically including:
-
Local Scale Learning Module: First, sampling is performed on the protein surface to obtain local fragments. For each point, the following features are extracted:
Atomic type features: $f_{atom} \in \mathbb{R}^{1\times6}$
Chemical descriptors: $f_{chem} \in \mathbb{R}^{1\times3}$
Geometric descriptors: $f_{geo} \in \mathbb{R}^{1\times3}$
Combining these features, the original feature vector for each point is represented as:
$f_i = [f_{atom}, f_{chem}, f_{geo}] \in \mathbb{R}^{1\times12}$
-
Global Scale Learning Module: Integrates sequence information and surface features, calculating attention weights:
$Attention(Q, K, V) = softmax(\frac{QK^T}{\sqrt{d_k}})V
2. Innovative Sampling Strategy
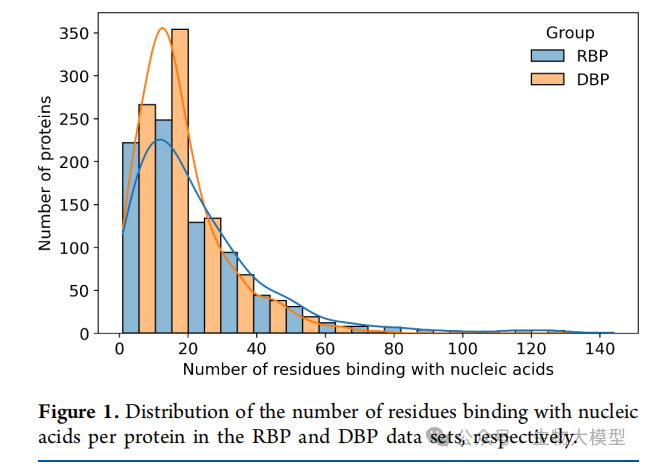
Figure 1 of the paper shows the distribution of the number of residues binding to nucleic acids for each protein in the RBP and DBP datasets, illustrating the complexity and scale of the binding interface.
The model employs a two-step sampling strategy:
-
Using the K-nearest neighbors algorithm to sample local fragments on the protein surface -
Choosing representative fragments through the farthest point sampling algorithm to ensure coverage of the entire protein surface
3. Multi-Modal Feature Fusion
The model innovatively combines the following features:
-
Geometric information of the protein surface -
Chemical properties -
Evolutionary conservation information -
Sequence features
Experimental Results and Performance Evaluation
1. Performance on Benchmark Datasets
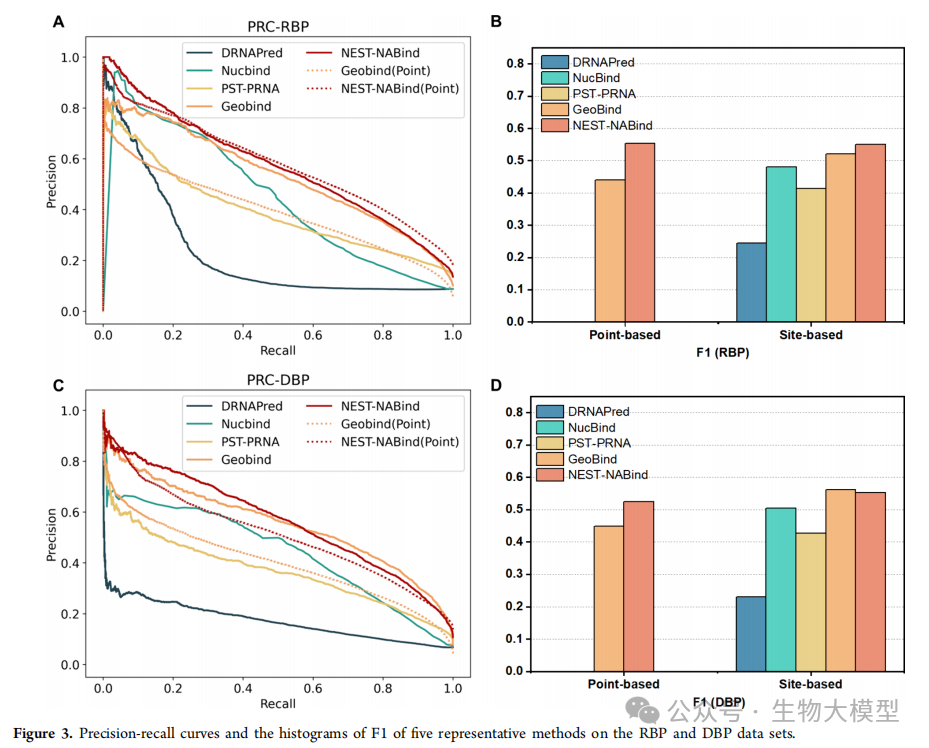
Figure 3 of the paper shows the precision-recall curves and F1 score histograms for different methods on the RBP and DBP datasets, clearly demonstrating the superior performance of NesT-NABind.
On the RNA-binding protein (RBP) dataset:
-
F1 score improved by 5.57% -
AUPRC improved by 3.64%
On the DNA-binding protein (DBP) dataset:
-
Point-level F1 score improved by 14.31% -
AUPRC improved by 21.83%
2. Large Protein Prediction Capability
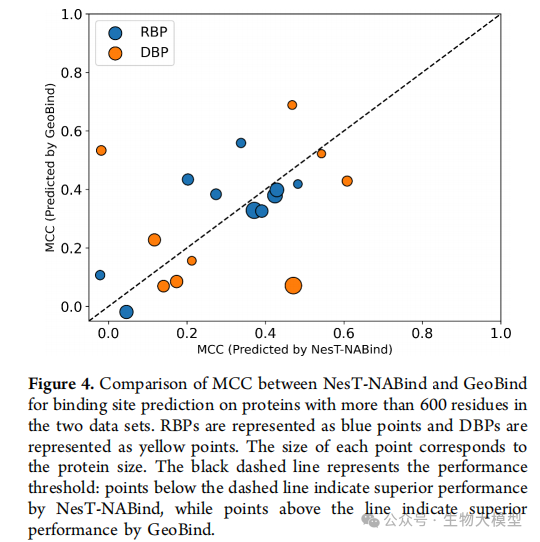
Figure 4 of the paper compares the performance of NesT-NABind and GeoBind in handling large proteins, where the size of the points indicates the scale of the proteins. The results show that NesT-NABind has a significant advantage when processing large proteins.
In proteins with more than 600 residues:
-
RBP dataset: superior performance in 60% of cases -
DBP dataset: superior performance in 66.67% of cases
3. Ablation Study Analysis
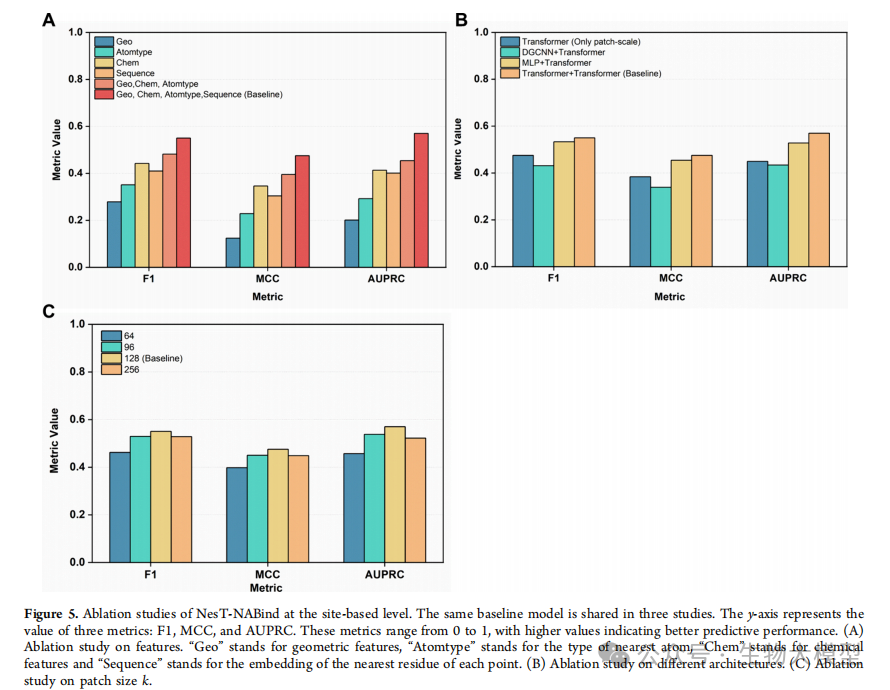
Figure 5 of the paper presents the results of three important ablation studies, validating the effectiveness of each component of the model:
-
Feature contribution analysis -
Module functionality validation -
Fragment size impact study
Case Analysis
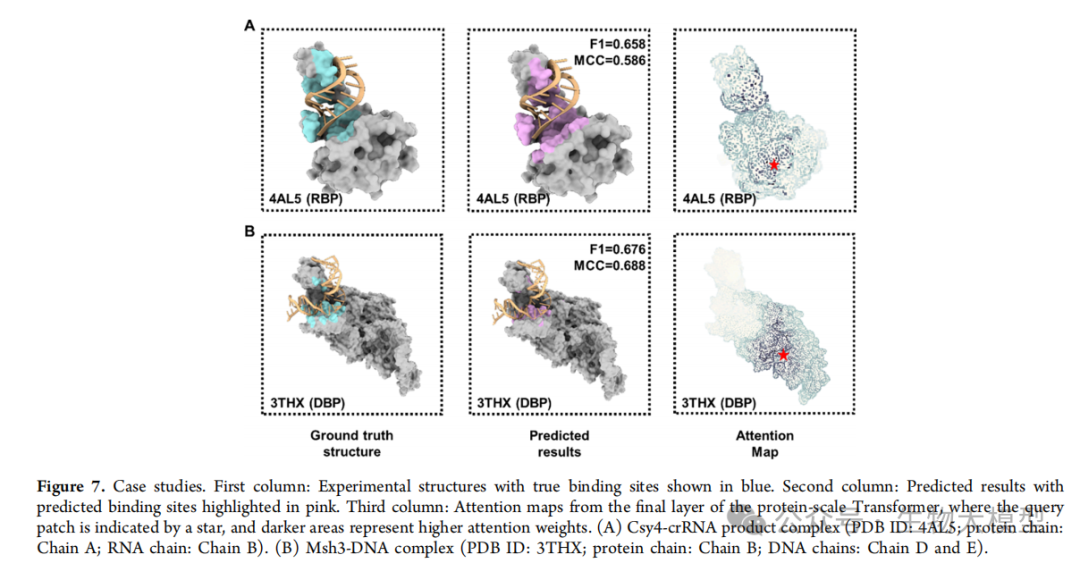
Figure 7 of the paper showcases two specific prediction cases:
-
CRISPR-associated nuclease Cas4 (PDB ID: 4AL5) -
DNA mismatch repair protein Msh3 (PDB ID: 3THX)
These cases clearly demonstrate the model’s accurate prediction capabilities across different types of proteins.
Technical Implementation and Open Source Code
Model implementation details:
-
Using the PyTorch deep learning framework -
Training environment: Single NVIDIA GeForce RTX 3060 GPU -
Training time: Approximately 3.5 hours for the RBP dataset, about 5 hours for the DBP dataset
The source code has been open-sourced on GitHub, available at: https://github.com/Xinyue-M/NesTNA-Bind
Conclusion and Outlook
NesT-NABind successfully addresses the limitations of traditional methods in predicting protein-nucleic acid binding sites through its innovative nested Transformer architecture. This method not only outperforms existing methods across various performance metrics but also demonstrates unique advantages in handling large proteins.
Future research directions include:
-
Expanding the training data scale -
Optimizing the Transformer architecture to handle longer input sequences -
Integrating prediction results with structural prediction models (such as AlphaFold3)
This research opens new avenues for computational predictions of protein-nucleic acid interactions and has significant application value for drug development and disease treatment.
Q&A Session: In-Depth Analysis of NesT-NABind Technology
Q1: How does the nested Transformer architecture in NesT-NABind work specifically? Why adopt this design?
In NesT-NABind, the nested Transformer architecture is designed to simultaneously capture local surface features and global protein information. This architecture consists of two main levels: local fragment scale and global protein scale.
At the local fragment scale, local fragments of size k are first sampled from the protein surface using the KNN algorithm. The features of each point in each fragment are represented as follows:
Each point’s feature vector includes atomic type, chemical properties, and geometric description:
These local fragments are then processed by a patch-scale Transformer, which includes a multi-head self-attention mechanism:
Where each head is calculated as:
At the global protein scale, another Transformer integrates the representations of all local fragments and sequence information to generate a global protein representation:
The advantage of this nested design is that it can simultaneously maintain local structural details and capture long-range dependencies, thereby better understanding the binding patterns on the protein surface.
Q2: How does NesT-NABind handle and integrate the geometric information of the protein surface? How are these features mathematically represented?
NesT-NABind employs a complex geometric feature extraction and representation system. First, the geometric features of the protein surface are primarily described through three key metrics:
-
Mean curvature (H):
-
Gaussian curvature (K):
-
Shape index (SI):
Where and are the principal curvatures. These geometric descriptors are normalized to the range [-1,1], and then concatenated with other features (such as atomic types and chemical properties) to form a complete point feature vector.
The system also quantifies the geometric complementarity of the protein surface through distance-related curvature, which is crucial for understanding the shape characteristics of binding sites.
Q3: How does NesT-NABind overcome the limitations of traditional methods when dealing with large proteins? What are the characteristics of its attention mechanism?
The advantage of NesT-NABind when handling large proteins primarily comes from its special attention computation mechanism. When processing large proteins, the calculation of attention scores follows the formula:
To handle long sequences, the model adopts a special hierarchical attention strategy:
-
Local attention computation:
-
Global attention integration:
This mechanism allows the model to effectively focus on relevant areas while maintaining computational efficiency when processing large proteins.
Q4: How does the feature fusion mechanism of NesT-NABind work? How are different types of features integrated?
NesT-NABind employs a complex multi-modal feature fusion mechanism, which primarily includes three levels of feature integration:
-
Point-level feature fusion:
-
Local fragment feature fusion:
-
Global feature fusion:
In the integration of sequence features, a pre-trained ESM model is used, and its output undergoes dimensional transformation:
Q5: How are the training strategy and loss function of the model designed? How is the sample imbalance issue addressed?
NesT-NABind employs a carefully designed training strategy. First, in the selection of the loss function, cross-entropy loss is used in conjunction with an adaptive learning rate adjustment mechanism. The learning rate update follows the strategy:
The initial learning rate is set to, and when the F1 score on the validation set does not improve for three consecutive epochs, the learning rate is adjusted as follows:
Until the learning rate reaches.
To address the sample imbalance issue, the model adopts the following strategies:
-
Point-based representation method to expand the dataset size, increasing the dataset size by about 17 times
-
Increase the proportion of positive samples:
-
RBP dataset: approximately 4% increase -
DBP dataset: approximately 3% increase
The training process of the model includes two key phases:
-
Local feature learning:
-
Global feature integration:
This training strategy not only effectively improves model performance but also significantly enhances the stability and accuracy of the model when handling large proteins. On the validation set, the model exhibits excellent generalization ability, especially when processing unbound conformations of proteins, achieving R² values of 0.82 and 0.60.
Q6: How does the surface fragment sampling pipeline of NesT-NABind work? Why is this sampling strategy crucial for model performance?
The surface fragment sampling pipeline is one of the key innovations of NesT-NABind. This process first calculates the solvent-excluded surface (SES) of the protein, using parameters of density 3.0 and a water probe radius of 1.5Å, and then normalizes it to a resolution of 1.2Å using PyMesh. The sampling process can be represented as:
First, for the coordinate representation of each point:
Then, using the KD-tree algorithm for nearest neighbor search, for each surface point:
The key of the sampling strategy lies in the two-layer sampling mechanism:
-
Local neighborhood sampling:
-
Global representative sampling:
The importance of this hierarchical sampling strategy is reflected in:
-
Ensuring the integrity of surface feature capture -
Reducing information redundancy -
Improving computational efficiency -
Maintaining the integrity of local structural information
Q7: How do the protein sequence learning module and surface feature learning module work together in NesT-NABind?
The collaboration between the sequence learning module and the surface feature learning module is achieved through a carefully designed feature alignment and fusion mechanism. Specifically:
Sequence encoding process:
Mapping sequence features to point features:
Feature fusion process:
In this process, key technical considerations include:
-
Dimension alignment: Transforming the 1280-dimensional ESM output through a linear layer to match the dimensions of surface features:
-
Spatial alignment: Aligning sequence features with surface features in space through the mapping relationship from points to residues:
Q8: How does NesT-NABind handle the chemical descriptors of protein surfaces? How do these descriptors affect the prediction of binding sites?
NesT-NABind employs a multi-level feature extraction and integration strategy when handling chemical descriptors of protein surfaces. First, for each surface point, three types of key chemical descriptors are extracted:
-
Hydrophobicity descriptor:
-
Protonation state:
-
Electrostatic potential:
These descriptors are integrated as follows:
In the prediction process, the influence of chemical descriptors is weighted through the attention mechanism:
The final site score is mapped to the residue level using the ScatterMax function:
Q9: How does the choice of patch size in NesT-NABind affect model performance? Why is 128 chosen as the optimal patch size?
The choice of patch size is a key parameter in model design that directly affects the effectiveness of feature extraction and computational efficiency. Through experiments, the model tested different patch sizes (k values): 64, 96, 128, and 256, corresponding to patch radii of approximately 6.56Å, 7.84Å, 8.89Å, and 12.12Å.
For a given patch size k, the feature extraction process can be represented as:
Subpatch sampling parameter settings:
Performance evaluation metrics calculation:
Experimental results show that when k=128 (corresponding to a radius of about 9Å), the model performance is optimal because:
-
This range is large enough to capture complete local structural information -
It does not introduce too much noise -
Computational complexity remains at a reasonable level
Q10: How does NesT-NABind perform when handling unbound conformational proteins? How does the model handle conformational changes?
NesT-NABind adopts special strategies when handling unbound conformational proteins. For each unbound conformational prediction, the model calculates a similarity score:
(RNA-binding protein) (DNA-binding protein)
Conformational changes are handled mainly through the following mechanisms:
-
Standardization of feature extraction:
-
Adjusting position encoding: Subtracting the coordinates of each patch center from the protein’s centroid coordinates:
-
Prediction score calculation:
Where t is the threshold determined based on the validation set. This handling approach allows the model to:
-
Adapt to conformational changes of proteins before and after binding -
Maintain prediction stability -
Capture key structural features -
Provide reliable binding site predictions
This strategy enables NesT-NABind to exhibit strong generalization ability when handling unbound conformations, particularly showing high prediction consistency when processing RNA-binding proteins.
NesTNA-Bind: Nested Transformer for Nucleic Acid Binding Site Prediction
Detailed Usage Guide
1. Environment Setup
First, configure the correct development environment:
# Clone the repository
git clone https://github.com/Xinyue-M/NesTNA-Bind.git
cd NesTNA-Bind
# Create virtual environment
conda create -n nestna python=3.8
conda activate nestna
# Install dependencies
pip install -r requirements.txt
The main dependencies include:
-
PyTorch >= 1.8.0 -
NumPy -
pandas -
scikit-learn -
Bio -
pymesh -
fair-esm
2. Data Preparation
2.1 Dataset Structure
data/
├── RBP/
│ ├── train/
│ │ ├── pdb_files/
│ │ ├── sequence_files/
│ └── test/
│ ├── pdb_files/
│ ├── sequence_files/
└── DBP/
├── train/
│ ├── pdb_files/
│ ├── sequence_files/
└── test/
├── pdb_files/
├── sequence_files/
2.2 Data Preprocessing
# Generate protein surface representation
python scripts/prepare_surface.py --input_dir data/RBP/train/pdb_files --output_dir data/RBP/train/surface_files
# Extract features
python scripts/extract_features.py --surface_dir data/RBP/train/surface_files --output_dir data/RBP/train/features
3. Model Training
# Train RBP dataset
python train.py \
--dataset rbp \
--train_dir data/RBP/train \
--val_dir data/RBP/val \
--model_dir checkpoints/rbp \
--batch_size 1 \
--epochs 30 \
--lr 1e-3
# Train DBP dataset
python train.py \
--dataset dbp \
--train_dir data/DBP/train \
--val_dir data/DBP/val \
--model_dir checkpoints/dbp \
--batch_size 1 \
--epochs 30 \
--lr 1e-3
4. Model Evaluation and Prediction
# Evaluate model performance
python evaluate.py \
--model_path checkpoints/rbp/best_model.pth \
--test_dir data/RBP/test \
--output_dir results/rbp
# Predict new proteins
python predict.py \
--model_path checkpoints/rbp/best_model.pth \
--input_pdb example.pdb \
--output_dir predictions
5. Important Parameter Descriptions
-
Surface sampling parameters:
-
<span>n = 2048</span>: Number of sampled patches -
<span>k = 128</span>: Number of points in each patch -
<span>n' = 16</span>: Number of subpatches -
<span>k' = 16</span>: Number of points in each subpatch -
Transformer parameters:
-
<span>num_blocks = 2</span>: Number of Transformer blocks -
<span>num_heads = 8</span>: Number of attention heads -
<span>d_local = 32</span>: Local feature dimension -
<span>d_seq = 32</span>: Sequence feature dimension -
<span>d_global = 64</span>: Global feature dimension
6. Result Visualization
# Generate prediction result visualization
python visualize.py \
--pdb_file example.pdb \
--prediction_file predictions/example_pred.txt \
--output_dir visualizations
7. Common Issues
-
Out of memory error
-
Reduce batch_size -
Decrease the number of sampled points -
Use gradient accumulation
CUDA error
-
Check GPU memory usage -
Update CUDA driver -
Try using the CPU version
Preprocessing failure
-
Ensure PDB file format is correct -
Check all dependencies are correctly installed -
View log files for detailed error information
8. Performance Optimization Suggestions
-
Data processing optimization:
# Use multi-processing to accelerate data preprocessing
python scripts/prepare_surface.py --num_workers 4
-
Training optimization:
# Use mixed precision training
python train.py --fp16 True
-
Prediction optimization:
# Batch prediction
python predict.py --batch_predict True
9. Citation Format
If you use NesTNA-Bind in your research, please cite:
@article{ma2024nest,
title={NesT-NABind: a Nested Transformer for Nucleic Acid-Binding Site Prediction on Protein Surface},
author={Ma, Xinyue and Li, Fenglei and Chen, Qianyu and Gao, Shenghua and Bai, Fang},
journal={Journal of Chemical Information and Modeling},
year={2024},
publisher={ACS Publications}
}
10. Contribution Guidelines
-
Fork this project -
Create a feature branch -
Commit changes -
Push to the branch -
Create a Pull Request
11. License
This project is licensed under the MIT License. See the LICENSE file for details.
12. Contact Information
If you have any questions, please contact us via:
-
Submitting an Issue -
Sending an email to the project maintainer
13. Changelog
v1.0.0 (2024-01-19)
-
Initial version release -
Implemented basic functionality -
Provided pre-trained model
v1.0.1 (2024-01-20)
-
Bug fixes -
Improved documentation -
Optimized performance
Stay tuned for project updates to get the latest features and improvements.

https://doi.org/10.1021/acs.jcim.4c01765
Training Fine-Tuning Applications of Biological Large Models&AI Protein Design Professional Tutorial Please enter Knowledge Planet 👇👇👇
(Customer service sssmd9 can issue invoices)