01
Training Background
In recent years, the use of high-performance computers for drug virtual screening has been widely applied. Computer-aided drug design can improve the success rate of drug development, reduce development costs, and shorten the development cycle, making it one of the core technologies in innovative drug research today.With the accumulation of medical big data and the development of artificial intelligence technology, the precise drug design using AI technology combined with big data is continuously promoting the development of innovative drugs. In the treatment plan for the novel coronavirus, a large class of drug molecules has been discovered through a series of computer-aided drug bio-computing methods that can effectively prevent the infection of the COVID-19 virus, providing new ideas for treating COVID-19.In response to the training needs of new and old customers, Beijing Soft Research International Information Technology Research Institute is holding a series of training classes on “Computer-Aided Drug Design Technology and Application Practice”. The training will be specifically organized by Interactive Education Technology Co., Ltd. (Beijing), and the relevant notifications are as follows://Click at the end to read the original text for the formal notification document
|
Topic One (Live) |
CADD Protein Structure Analysis, Virtual Screening, Molecular Docking (Protein-Protein, Protein-Peptide) |
|
Topic Two (Live) |
AMBER Molecular Dynamics Energy Optimization and Analysis, Binding Free Energy Calculation Topic |
|
Topic Three (Live) |
AIDD Artificial Intelligence (Shallow Learning and Deep Learning) Drug Discovery |
02
Training Features
 This series of courses consists of three topics, all adopting the online live broadcast format, providing software operation and case teaching unlimited replay videos, establishing a permanent course group, and long-term interactive Q&A. After completing the course, students can continue to communicate with professional teachers and classmates to consolidate learning content, thus better meeting the different aspects of students’ thesis and practical research work needs;
This series of courses consists of three topics, all adopting the online live broadcast format, providing software operation and case teaching unlimited replay videos, establishing a permanent course group, and long-term interactive Q&A. After completing the course, students can continue to communicate with professional teachers and classmates to consolidate learning content, thus better meeting the different aspects of students’ thesis and practical research work needs;
Topic One
Will guide you step-by-step to practically learn protein structure analysis, homology modeling, virtual screening, molecular docking (semi-flexible, flexible docking, protein-protein, protein-peptide, metal enzyme protein-ligand, nucleic acid-small molecule, covalent docking, etc.), pharmacophore modeling, quantitative structure-activity relationship, fragment-based drug design, Gromacs molecular dynamics simulation and result analysis, focusing on case explanations and exercises to achieve immediate application effectiveness, helping students systematically master computer-aided drug design technology and assist academic research;
Topic Two
Starting from the installation and operation of AMBER, in-depth study of the acquisition and construction of object models, energy optimization, molecular dynamics simulation, trajectory feature acquisition, and analysis of interaction mechanisms based on energy, and reproducing classic literature.
Topic Three
Through classification and regression tasks of machine learning and deep learning, molecular features, model evaluation, parameter optimization, and model selection, shallow learning classification for virtual screening, using ensemble machine learning methods, DNN, GCN, GAT, and other mainstream deep learning model practices, as well as practical applications combined with related topics, truly bringing you into the field of artificial intelligence drug discovery.
☟ Swipe up and down to see more
Slide for more photos
☟ Swipe up and down to see more
Slide for more photos
03
Training Time
CADD Protein Structure Analysis, Virtual Screening, Molecular Docking (Protein-Protein, Protein-Peptide)
June 24-26, 2022
Online Live (Three Days of Classes)
July 1-3, 2022
Online Live (Three Days of Classes)
AMBER Molecular Dynamics Energy Optimization and Analysis, Binding Free Energy Calculation Topic
July 2-5, 2022
Online Live (Four Days of Classes)
AIDD Artificial Intelligence (Shallow Learning and Deep Learning) Drug Discovery Topic
June 24-26, 2022
Online Live (Three Days of Classes)
July 2-3, 2022
Online Live (Two Days of Classes)
04
Training Outline
CADD Protein Structure Analysis, Virtual Screening, Molecular Docking
(Protein-Protein, Protein-Peptide)
|
Time |
Course Name |
Course Content |
|
Day One Morning |
Basics of Biomolecular Interactions |
1. Methods for studying biomolecular interactions 1.1 Principles of protein-small molecule and protein-protein interactions 1.2 Molecular docking to study biomolecular interactions 1.3 Protein-protein docking to study molecular interactions |
|
Protein Database |
1. Introduction to the PDB database 1.1 Functions of the PDB protein database 1.2 Resources for obtaining PDB protein data 1.3 Importance of the PDB protein database for drug development 2. Using the PDB database 2.1 Types of target protein structures, data interpretation and download 2.2 Downloading target protein structure sequences 2.3 Background analysis of target proteins 2.4 Ways to obtain related data resources 2.4 Batch download of protein crystal structures |
|
|
Day One Afternoon |
Protein Structure Analysis |
1. Introduction to Pymol software 1.1 Software installation and initial settings 1.2 Basic knowledge introduction (such as hydrogen bonds, etc.) 2. Using Pymol software 2.1 Diagrams of protein-small molecule interactions 2.2 Diagrams of protein-protein interactions 2.3 Surface diagrams of proteins and small molecules, electrostatic potential representations 2.4 Superimposing and comparing protein and small molecule structures 2.5 Drawing interaction forces 2.6 Making Pymol animations Example explanations and exercises: (1) Interaction of nilotinib with the target, listing the interacting amino acids and exporting the binding mode diagram (2) Making a binding pocket surface diagram (3) Superimposing the PDB structure of the Bcr/Abl target (4) Making a protein interaction animation (5) For the protein crystal complex of ACE2 and the COVID-19 Spike protein, making a protein-protein interaction |
|
Morning of Day Two |
Homology Modeling |
1. Introduction to the principles of homology modeling 1.1 Functions and usage scenarios of homology modeling 1.2 Methods of homology modeling 2. Swiss-Model homology modeling; 2.1 Searching for homologous proteins (using methods like blast) 2.2 Protein sequence alignment 2.3 Selecting protein templates 2.4 Building protein models 2.5 Model evaluation (protein Ramachandran plot) 2.6 Protein model optimization Example explanations and exercises: Modeling using the 2019-nCoV spike protein sequence, evaluating the model based on corresponding parameters and methods |
|
Afternoon of Day Two |
Small Molecule Construction |
1. Introduction to ChemDraw software 1.1 Constructing small molecule structures 1.2 Calculating the physicochemical properties of small molecules (such as molecular weight, clogP, etc.) Example explanations and exercises: Constructing macrocyclic compounds, amino acids, DNA, RNA, etc. |
|
Small Molecule Compound Library
|
1. Small molecule databases 1.1 Introduction and usage of databases like DrugBank, ZINC, ChEMBL 1.2 Introduction and usage of natural product and traditional Chinese medicine databases |
|
|
Morning of Day Three |
Biomolecular Interactions I |
1. Basics of molecular docking 1.1 Principles of molecular docking and introduction to docking software 2. Using molecular docking software (Autodock) 2.1 Semi-flexible docking 2.1.1 Preparing small molecule ligands for optimization 2.1.2 Optimizing protein receptors and preparing coordinate files 2.1.3 Calculating grid points for protein receptors 2.1.4 Performing semi-flexible docking calculations 2.2 Evaluating docking results 2.2.1 Comparing crystal structure conformations 2.2.2 Evaluating docking results from an energy perspective 2.2.3 Evaluating docking results through clustering analysis 2.2.4 Selecting the optimal binding conformation 2.2.5 Comparing docking results with known active compounds Example explanations and exercises: Semi-flexible docking of kinase Bcr/Abl target inhibitors |
|
Afternoon of Day Three |
Biomolecular Interactions II
|
2.3 Flexible docking 2.3.1 Preparing small molecule ligands for optimization 2.3.2 Optimizing protein receptors and preparing coordinate files 2.3.3 Calculating grid points for protein receptors 2.3.4 Performing flexible docking calculations and evaluating results 2.3.6 Comparing and selecting between semi-flexible and flexible docking Example explanations and exercises: Flexible docking of Bcr/Abl target inhibitors |
|
Virtual Screening |
3. Using molecular docking for virtual screening (Autodock) 3.1 Defining virtual screening, building processes, and demonstrations 3.2 Selecting target proteins and obtaining compound libraries 3.3 Conducting virtual screening 3.4 Analyzing results (scoring values, energy, and interaction analysis) Example explanations and exercises: Virtual screening of Bcr/Abl target inhibitors |
|
|
Small Molecule Format Conversion |
1. Introduction and usage of openbabel 1.1 Introduction to openbabel software 1.2 Types of small molecule structures 1.3 Converting types of small molecule structures |
|
| Q&A | Answering questions related to the first three days of learning | |
|
Morning of Day Four |
Expanding docking usage scenarios (Part I) |
1. Protein-protein macromolecular docking 1.1 Application scenarios of protein-protein docking 1.2 Introduction to related programs 1.3 Preparing receptors and ligands for optimization 1.4 Loading receptor and ligand molecules 1.5 Setting docking sites for protein-protein interactions 1.6 Analyzing and interpreting results from protein-protein docking Example explanations and exercises: Docking of COVID-19 Spike protein and host protein ACE2 2. Protein-peptide docking 2.1 Introduction to protein-peptide interactions 2.2 Preprocessing protein-peptide molecules 2.3 Docking protein-peptide molecules 2.4 Displaying and analyzing docking results Example explanations and exercises: Docking of COVID-19 target 3CL with peptide/class-peptide inhibitors |
|
Afternoon of Day Four |
Expanding docking usage scenarios (Part II) |
4. Small molecule docking with small molecules 4.1 Introduction to small molecule-small molecule interactions 4.2 Preprocessing small molecule structures 4.3 Docking small molecules with small molecules 4.4 Displaying and analyzing docking results Example explanations and exercises: Docking of cyclodextrin with drug small molecules 5. Nucleic acid-small molecule docking 5.1 Application scenarios of nucleic acid-small molecule interactions 5.2 Introduction to nucleic acid-small molecule interactions 5.3 Preprocessing nucleic acid-small molecules 5.4 Docking nucleic acid-small molecules 5.5 Displaying and analyzing related results Example explanations and exercises: Docking of DNA G-quadruplex and ligand molecules |
|
Covalent Docking |
6. Principles and application scenarios of covalent docking 6.2 Preprocessing proteins and covalently binding ligands 6.3 Covalent docking of drug molecules with target proteins 6.6 Displaying and analyzing related results Example explanations and exercises: Covalent docking of kinase target EGFR inhibitors |
|
|
Morning of Day Five |
Fragment-Based Drug Design |
1. Fragment-based drug design and discovery 1.1 Constructing fragment-based compound libraries 1.2 Skeleton replacement 1.3 Fragment linking 1.4 Fragment growth 1.5 Generating compound libraries based on pharmacophores 1.6 Generating compound libraries based on protein binding pockets 1.7 Generating compound libraries based on molecular descriptors 1.8 Constructing compound libraries based on BREED rules 1.9 Screening compound libraries based on fragments Example explanations and exercises: Optimization and modification of Bcr/Abl target inhibitors based on fragments |
|
Afternoon of Day Five |
Structure-Activity Relationship Analysis |
1. Building 3D-QSAR models (using Sybyl software) 1.1 Constructing small molecules 1.2 Creating a small molecule database 1.3 Adding charges and optimizing energy of small molecules 1.4 Determining and superimposing active conformations of molecules 1.5 Creating 3D-QSAR models 1.6 Building CoMFA and CoMSIA models 1.7 Validating models with test sets 1.8 Analyzing model parameters 1.9 Analyzing model isopotential maps 1.10 Guiding drug design with 3D-QSAR models Example explanations and exercises: Building structure-activity relationship models and predicting activity for kinase target Bcr/Abl inhibitors |
|
All Day on Day Six |
Molecular Dynamics Simulation |
1. Introduction to molecular dynamics (using GROMACS software) 1.1 Basic principles of molecular dynamics 1.2 Introduction to the Linux system 1.3 Introduction to molecular dynamics software (Gromacs) 2. Conducting molecular dynamics simulations with Gromacs 2.1 Processing ligand molecules 2.2 Processing protein structures 2.3 Modifying protein coordinate files 2.4 Modifying topology files 2.5 Constructing boxes and adding solvents 2.6 Balancing system charge 2.7 Energy minimization 2.8 NVT balancing 2.9 NPT balancing 2.10 Outputting molecular dynamics simulations 3. Analyzing molecular dynamics results 3.1 Observing trajectory files 3.2 Plotting energy data 3.3 Processing trajectory corrections 3.4 Analyzing radius of gyration 3.5 Calculating RMSD changes of protein conformations 3.6 Calculating RMSF changes of atomic positions 3.7 Clustering protein-ligand conformations 3.8 Analyzing hydrogen bond interactions between proteins and ligands 3.9 Analyzing interaction energies between proteins and ligands Example explanations and exercises: (1) Simulating pure proteins of lysozyme in water (2) Simulating complexes of T4 lysozyme and ligands |
|
Q&A |
Answering questions related to the last three days of learning |
AMBER Molecular Dynamics Simulation Energy Optimization, Binding Free Energy Calculation Topic
|
Day One |
||
|
Time |
Course Content |
Main Knowledge Points |
|
AM 9:00~9:50 |
1. Introduction to Molecular Dynamics Teaching Objective: Understand the content in this direction, theoretical basis, and research significance. |
1 Introduction to Molecular Mechanics
1.1 Basic assumptions of molecular mechanics 1.2 Main forms of molecular mechanics 2 Molecular Force Fields 2.1 Introduction to molecular force fields 2.2 Principles of molecular force fields 2.3 Classification and applications of molecular force fields |
|
AM 10:00~10:50 |
2. Introduction to LINUX Teaching Objective: Master the numerical computation platform, familiarize with computer languages, and be able to use the vim editor to edit files simply. |
3 Introduction to LINUX 3.1 User groups and permissions3.2 Directory file attributes3.3 Basic LINUX commands3.4 LINUX environment variables3.5 Common shell command exercises |
|
AM 11:00~12:00 |
||
|
PM 14:00~14:50 |
3. Introduction and Installation of AMBER Teaching Objective: Understand the historical development of Amber software, familiarize with installation environments, and support environment compilation. |
4 Introduction and Installation of AMBER 4.1 Introduction and installation of GCC4.2 Introduction and installation of Open MPI4.3 Running AMBER installation4.4 LINUX operation exercises |
|
PM 15:00~15:50 |
||
|
PM 16:00~17:00 |
||
|
Day Two |
||
|
Time |
Course Content |
Main Knowledge Points |
|
AM 9:00~9:50 |
4. Obtaining Research Object Models Teaching Objective: How to establish research objects, familiarize with the usage of protein databases, and how to model research objects. |
5 Preprocessing Model Files 5.1 Introduction to model sources5.2 Introduction to protein files |
|
AM 10:00~10:50 |
5. Constructing Research Object Models Teaching Objective: Familiarize with the preprocessing workflow of models, master the writing of input files, and be able to independently complete the preparatory work before system dynamics. |
6 Preprocessing Model Files 6.1 Protein preprocessing6.2 Small molecule preprocessing |
|
AM 11:00~12:00 |
6.3 Introduction to AMBER force fields 6.4 Introduction to topology files and coordinate files 6.5 Generating top and crd files 6.6 Using the tleap module Case Practice: Ø Preprocessing of HIV-1 complex |
|
|
PM 14:00~14:50 |
6. Molecular Dynamics Simulation Teaching Objective: Molecular dynamics workflow, principles of AMBER software dynamics, and completing operational exercises for molecular dynamics simulations. |
7 Energy optimization and molecular dynamics simulation
7.1 Significance and methods of energy optimization 7.2 Significance and methods of adjusting simulation temperature 7.3 Classification and selection of solvent models 7.4 Writing input files for dynamics simulation 7.5 Running molecular dynamics simulations 7.6 Interpreting output content 7.7 Exercises and Q&A Case Practice: Ø Energy optimization and molecular dynamics simulation of HIV-1 complex |
|
PM 15:00~15:50 |
||
|
AM 16:00~17:00 |
||
|
Day Three |
||
|
Time |
Course Content |
Main Knowledge Points |
|
AM 9:00~9:50 |
7. Calculating Binding Free Energy Teaching Objective: Familiarize with the significance of binding free energy calculations, MMPBSA methods, and processes. |
8 Enthalpy Change Calculations 8.1 Analyzing and retrieving experimental data8.2 Principles of MM/PBSA binding free energy calculations8.3 Explanation and classification of GB models8.4 Writing input files for enthalpy change8.5 Interpreting enthalpy change results |
|
AM 10:00~10:50 |
9 Entropy Change Calculations 9.1 Principles of Nmode entropy change calculations9.2 Writing input files for entropy change9.3 Interpreting entropy change results9.4 Comparing experimental and theoretical values Case Practice: Ø Binding free energy calculations between HIV-1 and inhibitors |
|
|
AM 11:00~12:00 |
||
|
PM 14:00~14:50 |
8. Visualization Software Teaching Objective: Familiarize with the channels for obtaining visualization software, installation, and basic usage, using visualization software to assist in research work. |
10 3D Visualization Analysis 10.1 Installing and using VMD10.2 Installing and using Pymol |
|
PM 15:00~15:50 |
9. Obtaining Trajectory Features Based on Molecular Dynamics Teaching Objective: Starting from conformations obtained from dynamics simulations, gain insights into conformational transitions, explain experimental imaginations, and predict experimental results. |
11 Conformational Analysis 11.1 RMSD analysis11.2 B-Factory analysis11.3 RMSF analysis |
|
AM 16:00~17:00 |
11.4 RG analysis11.5 VMD animation display11.6 Distance and angle measurements11.7 Solvent Accessible Surface Area (SASA) | |
|
Day Four |
||
|
Time |
Course Content |
Main Knowledge Points |
|
AM 9:00~9:50 |
10. Analyzing Interaction Mechanisms Based on Energy Teaching Objective: From an energy perspective, analyze interaction mechanisms between molecules, residues, and important groups, providing theoretical guidance for experiments. |
12 Energy Analysis 12.1 Residue decomposition (interaction analysis)12.2 Alanine scanning (finding hotspot residues)12.3 Hydrogen bond networks (salt bridges, pi-pi conjugation, and other interactions)12.4 Exercises and Q&A |
|
AM 10:00~10:50 |
||
|
AM 11:00~12:00 |
||
|
PM 14:00~14:50 |
11. Reproducing Classic Works Teaching Objective: Guide beginners to understand classic works in this direction, reproduce important analysis methods in the work, and deepen students’ understanding of this direction. |
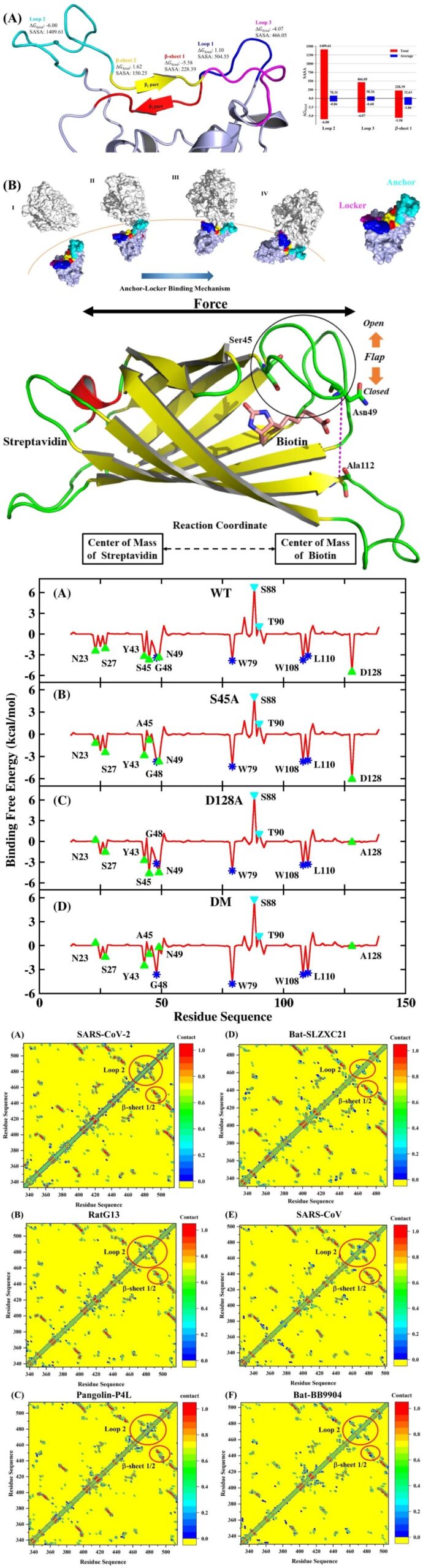
13 Reproducing Classic Literature Works (Students are required to download and read carefully before class) 13.1 Nanoscale 2020, 12, 7134−7145. (a) RMSD and 2D_RMSD testing the stability of simulations(b) Comparing and analyzing binding free energy(c) Thermodynamic integration to calculate relative binding free energy(d) Analyzing hydrogen bond networks(e) Residue decomposition predicting hotspot amino acids 13.2 J. Chem. Inf. Model. 2021, 61, 7, 3529–3542 (a) Alanine scanning predicting hotspot amino acids(b) Residue contact analysis(c) Analyzing temperature factors (B-factor)(d) Analyzing solvent accessible surface area(e) Reproducing dissociation mechanisms through umbrella sampling dynamics 13.3 Exercises and Q&A |
|
PM 15:00~15:50 |
||
|
PM 16:00~17:00 |
AIDD Artificial Intelligence (Shallow Learning and Deep Learning) Drug Discovery
|
Time |
Course Content |
Course Content |
|
First Morning |
Computer-Aided Drug Molecular Design |
1. Introduction to Computer-Aided Drug Design (CADD) 2. Basic methods of CADD 2.1 Molecular Docking 2.2 Pharmacophore 2.3 QSAR and QSPR 2.4 Introduction to various pharmaceutical research databases |
|
First Afternoon |
Installation and Configuration of Anaconda3 |
3. Anaconda3 3.1 Pandas 3.2 NumPy 3.3 RDKit 3.4 scikit-learn 3.5 Pytorch 3.6 Tensorflow 3.7 DeepChem 3.8 XGBoost |
|
Second Morning |
Introduction to AIDD —— Classification and Regression Tasks |
1. Building and Applying Classification Models 1.1 Principles of Logistic Regression Algorithm 1.2 Principles of Naive Bayes Algorithm 1.3 Principles of k-Nearest Neighbors Algorithm 1.4 Principles of Support Vector Machine Algorithm 1.5 Principles of Random Forest Algorithm 1.6 Principles of Gradient Boosting Algorithm 2. Introduction to Molecular Features 2.1 Molecular Descriptors 2.2 Molecular Fingerprints 2.3 Molecular Graphs |
|
Second Afternoon |
Drug Discovery Based on Shallow Machine Learning (Goal: Guide students to implement toxicity prediction models based on other three algorithms like KNN, SVM, XGBoost, and use them for predicting the toxicity of small molecule compounds) |
3. Model Evaluation Methods 3.1 Cross-validation 3.2 External validation 3.3 Common evaluation metrics for classification models 3.4 Confusion matrix 3.5 Accuracy 3.6 Sensitivity 3.7 Specificity 4. Parameter Optimization and Model Selection 4.1 Hyperparameter optimization 4.2 Criteria for model selection Model example explanations and exercises (literature reproduction): Ø Using a given dataset as an example, building and using a toxicity prediction model related to CYPs inhibitors based on the random forest algorithm |
|
Third Morning |
Drug Discovery Based on Shallow Learning—Regression Tasks
|
1. Building and Applying Regression Models 2. Common Evaluation Metrics for Regression Models 2.1 MSE 2.2 MAE 2.3 R2 3. Model Selection 3.1 Hyperparameter Optimization 3.2 Selecting the Optimal Model |
|
Third Afternoon |
Virtual Screening Based on Shallow Learning Classification (Goal: Guide students to implement pIC50 value prediction models based on other three algorithms and use them for predicting pIC50 values of small molecule compounds) |
Model example explanations and exercises (literature reproduction): Ø Using tubulin as an example, the practical role of machine learning classification tasks in drug discovery Ø Using a given dataset as an example, building and using a pIC50 value prediction model based on the random forest algorithm |
|
Fourth Day |
Drug Discovery Based on Deep Learning |
1. Development History of Deep Learning 1.1 Applications of Deep Learning in Drug Development 1.2 DNN 1.3 GCN 1.4 GAT 1.5 KGCN 1.6 FP-GNN 2. Common Frameworks for Deep Neural Networks 2.1 PyTorch 2.2 TensorFlow 3. Introduction and Usage of DEEPCHEM Model example explanations and exercises (literature reproduction): Ø Integrated machine learning methods of DEEPCHEM and how to load and use them |
|
Fifth Day |
Practical operations using mainstream deep learning models such as DNN, GCN, GAT |
Example explanations and exercises (literature reproduction): Ø Using a given dataset as an example, predicting small molecule anti-breast cancer activity using mainstream deep learning models Ø Basic ideas, logic, and topic design for breast cancer prediction |
Some Case Illustrations:
05
Training Instructors
 CADD Topic Instructor: Lectured by associate professors from key national universities and key construction pharmaceutical colleges under the national “Double First-Class” and “211 Project”, master’s supervisors. Published over 20 SCI research papers and hosted or participated in multiple national and provincial natural science foundation projects. With years of experience in new drug molecular design and development, mainly proficient in drug design methods such as CADD and AIDD.AMBER Topic Instructor: A specially appointed professor from a key university in Shandong Province and a member of the provincial youth team, whose research team has published over 50 SCI papers in internationally renowned journals like “J. Am. Chem. Soc.”, “Phys. Chem. Chem. Phys.”, “Nanoscale”, “J. Chem. Phys.”. Two papers published as the first author in JACS belong to top-tier journals in this field (一区论文,IF: 14.612). Hosted multiple national natural science foundation and provincial-level projects. Mainly engaged in research on biomolecular quantification calculations and molecular dynamics simulations, accumulating rich experience in dynamics simulations, quantification calculations, and binding free energy calculations.AIDD Topic Instructor: Lectured by associate professors from key national universities and key construction colleges under the national “Double First-Class” and “211 Project”, master’s supervisors. In the past five years, published over 10 representative papers, 8 patents, 8 software copyrights, and hosted or participated in over 10 national and provincial natural science foundation projects. With years of experience in the design, synthesis, and activity research of anti-cancer drug molecules, mainly proficient in drug design methods such as bioinformatics, artificial intelligence (deep learning), and data mining.
CADD Topic Instructor: Lectured by associate professors from key national universities and key construction pharmaceutical colleges under the national “Double First-Class” and “211 Project”, master’s supervisors. Published over 20 SCI research papers and hosted or participated in multiple national and provincial natural science foundation projects. With years of experience in new drug molecular design and development, mainly proficient in drug design methods such as CADD and AIDD.AMBER Topic Instructor: A specially appointed professor from a key university in Shandong Province and a member of the provincial youth team, whose research team has published over 50 SCI papers in internationally renowned journals like “J. Am. Chem. Soc.”, “Phys. Chem. Chem. Phys.”, “Nanoscale”, “J. Chem. Phys.”. Two papers published as the first author in JACS belong to top-tier journals in this field (一区论文,IF: 14.612). Hosted multiple national natural science foundation and provincial-level projects. Mainly engaged in research on biomolecular quantification calculations and molecular dynamics simulations, accumulating rich experience in dynamics simulations, quantification calculations, and binding free energy calculations.AIDD Topic Instructor: Lectured by associate professors from key national universities and key construction colleges under the national “Double First-Class” and “211 Project”, master’s supervisors. In the past five years, published over 10 representative papers, 8 patents, 8 software copyrights, and hosted or participated in over 10 national and provincial natural science foundation projects. With years of experience in the design, synthesis, and activity research of anti-cancer drug molecules, mainly proficient in drug design methods such as bioinformatics, artificial intelligence (deep learning), and data mining.
06
Training Fees
(Including registration fee, training fee, and material fee)
| Course Name | Public Price (Yuan) | Self-Paid Price (Yuan) |
| CADD Topic | 5600 | 5200 |
| AMBER Topic | 3900 | 3500 |
| AIDD Topic | 5600 | 5200 |
Fees will be provided for reimbursement with official machine-printed invoices and paper notification documents stamped with official seals; if you need to issue a conference fee, please contact the enrollment teacher for a conference invitation letter;
07
Value-Added Services
1. All registered students will receive the training textbook for preview and electronic materials during the class;2. Provide unlimited replay videos: registered students will receive videos of software operations for the learned topics after the course ends;3. Price Discounts:Discount One: A discount of 400 yuan is available for any topic registered and paid before June 6, 2022 (limited to the first eight); Discount Two: An additional discount is available for the same person registering for two topic courses (please consult the enrollment contact for specifics)4. Questions raised by students can receive long-term answers and guidance from teachers after the course ends; 5. After the course ends, students will receive a professional skills completion certificate issued by Beijing Soft Research International Information Technology Research Institute Training Center;
08
Contact Information

09
Course Q&A
How to register and pay?
1. Call the teacher responsible for administrative enrollment to register
2. Payment supports public-to-public transfer, personal advance payment (public funds will be refunded promptly after payment, and a payment proof can be issued)

Currently, over 10,000 people have joined us

