First Author: Zhu Huaxin, Hao Zheng
Corresponding Author: Li Meihua
Affiliation: Department of Neurosurgery, First Affiliated Hospital of Nanchang University
[REF: Zhu H, Hao Z, Xing Z, Tan J, Zhao Y, Li M. Impinging Flow Induces Expression of Monocyte Chemoattractant Protein-1 in Endothelial Cells Through Activation of the c-Jun N-terminal Kinase/c-Jun/p38/c-Fos Pathway [published online ahead of print, 2022 May 14]. World Neurosurg. 2022;S1878-8750(22)00663-5. doi:10.1016/j.wneu.2022.05.032] PMID: 35580782
Abstract
Introduction
Intracranial aneurysm (IA) is one of the most common cerebrovascular diseases, with an incidence of approximately 1-3% in the general population[50]. IA typically refers to the abnormal bulging of the arterial wall. After rupture, it can cause intracranial hemorrhage and hematoma due to mass effect, leading to complications such as intracranial vasospasm, brain tissue damage, and cerebral edema, which severely threaten the life and health of patients[17,56]. IA is currently believed to be related to several factors, including hemodynamic changes, age, genetics, hypertension, and environment[31]. The mechanisms underlying IA formation require further in-depth research.
Methods
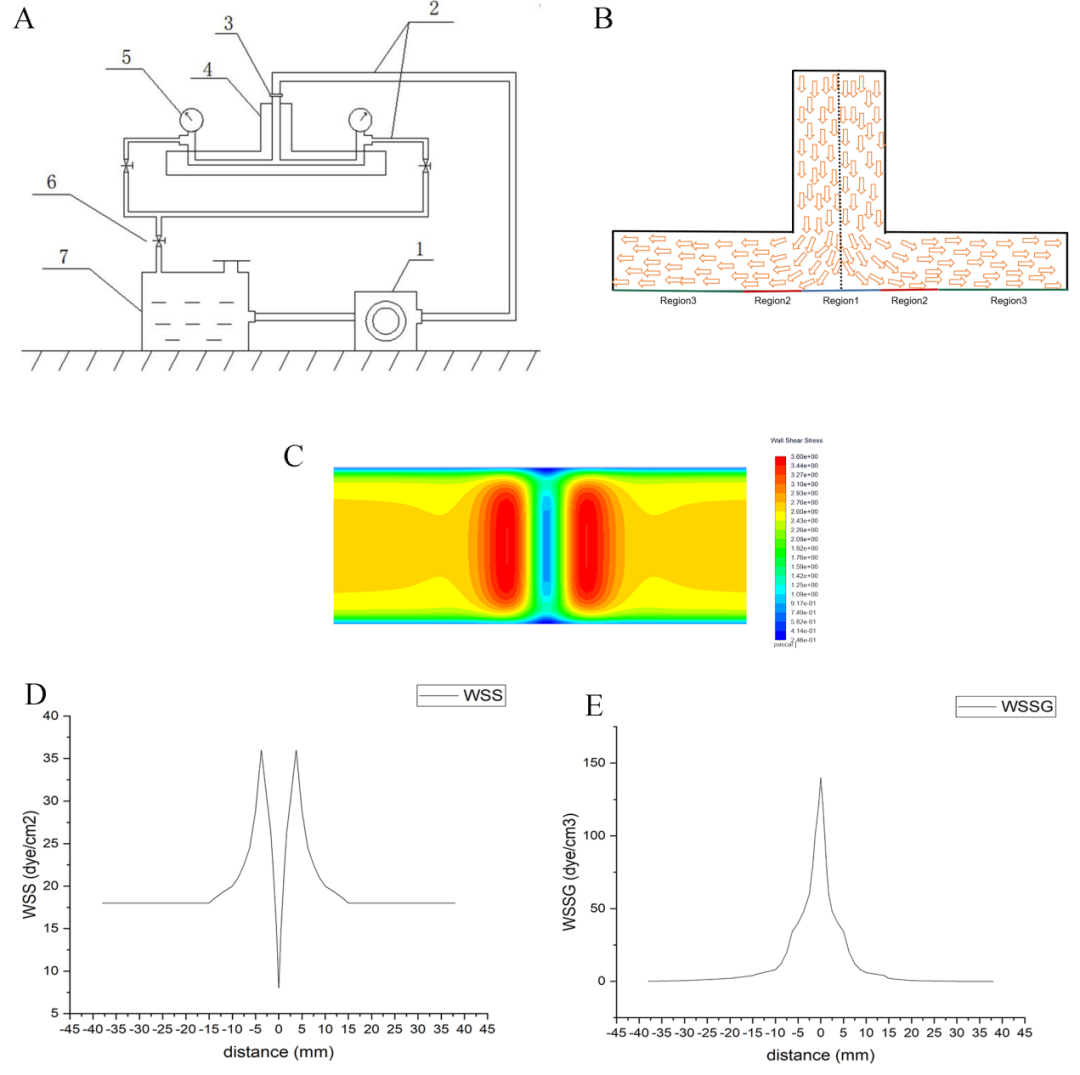
Modified T-shaped Flow Chamber
In this study, a modified T-shaped flow chamber was used to simulate fluid flow at arterial bifurcations, as well as WSS and wall shear stress gradient (WSSG)[24,60-62]. Figure 1A is a schematic diagram of the improved T-shaped flow chamber. Fluent software (Ansys, Canonsburg, PA, USA) was used to simulate the flow chamber, flow rate, WSS, WSSG, and turbulence intensity. The flow rate used in our experiments was 500 ml/min, with WSS ranging from 18-36 dynes/cm² and WSSG from 0-140 dynes/cm³ (Figure 1D, E). The experimental device operated at 37°C, with impinging flow acting for 0, 3, or 6 hours. Based on the distribution of WSS, human umbilical vein endothelial cells (HUVECs) on the glass slides were divided into three different regions. Region 1 was subjected to low to moderate WSS and abrupt WSSG, Region 2 to high WSS and abrupt WSSG, and Region 3 to moderate WSS and low, stable WSSG (Figure 1B, C). HUVECs not subjected to impinging flow served as the static control group.
Cell Culture
Western Blot Analysis (WB)
All three regions of HUVECs were collected and prepared for Western blot (WB) analysis. Cells were lysed in RIPA buffer containing phenylmethylsulfonyl fluoride (PMSF) and protein phosphatase inhibitors (Beijing Solarbio Technology Co., Ltd.) at 4°C for 30 minutes. The cell lysates were centrifuged at 14,000×g at 4°C for 15 minutes, and the supernatant was collected. The protein concentration of each sample was determined using a BCA protein assay kit (Beijing Solarbio Technology Co., Ltd.). Cell lysates were mixed with SDS-PAGE loading buffer (Beijing Solarbio Science & Technology) in equal proportions and heated in a water bath at 100°C for 5-10 minutes to denature the proteins. After cooling to room temperature, the mixture was centrifuged at 12,000×g for 2-5 minutes. Denatured proteins were analyzed by SDS-PAGE and transferred to a polyvinylidene fluoride membrane for immunoreaction. The membrane was incubated for 1 hour in blocking solution (5% milk and 0.1% Tween20 in Tris-buffered saline), followed by overnight incubation at 4°C with primary antibodies (ERK [ab184699] (1:1000), p-ERK [ab201015] (1:1000), JNK [ab179461] (1:1000), p-JNK [ab124956] (1:1000), p38 [ab170099] (1:2000), p-p38 [ab195049] (1:1000), c-Fos [ab184666] (1:1000), c-Jun [ab32137] (1:1000), MCP-1 [ab214819] (1:1000), and β-actin [ab8226] (1:1000); all purchased from Abcam). The membrane was washed three times (5 minutes each) with TBS containing 0.1% Tween20 and incubated at room temperature for 2 hours with secondary antibodies (1:2000) (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Protein blots were detected using the Pierce ECL Plus Western Blotting Kit (ThermoFisher Scientific, Waltham, MA, USA).
Gene Expression Analysis
Statistical Analysis
Results are expressed as mean ± standard error. Data were analyzed using SPSS software (version 19.0; IBM). The statistical significance of differences between groups was assessed using one-way ANOVA, while the chi-square test was used for categorical variables. A P-value < 0.05 was considered statistically significant. Each experiment was performed in triplicate to ensure the overall accuracy of the data.
Results
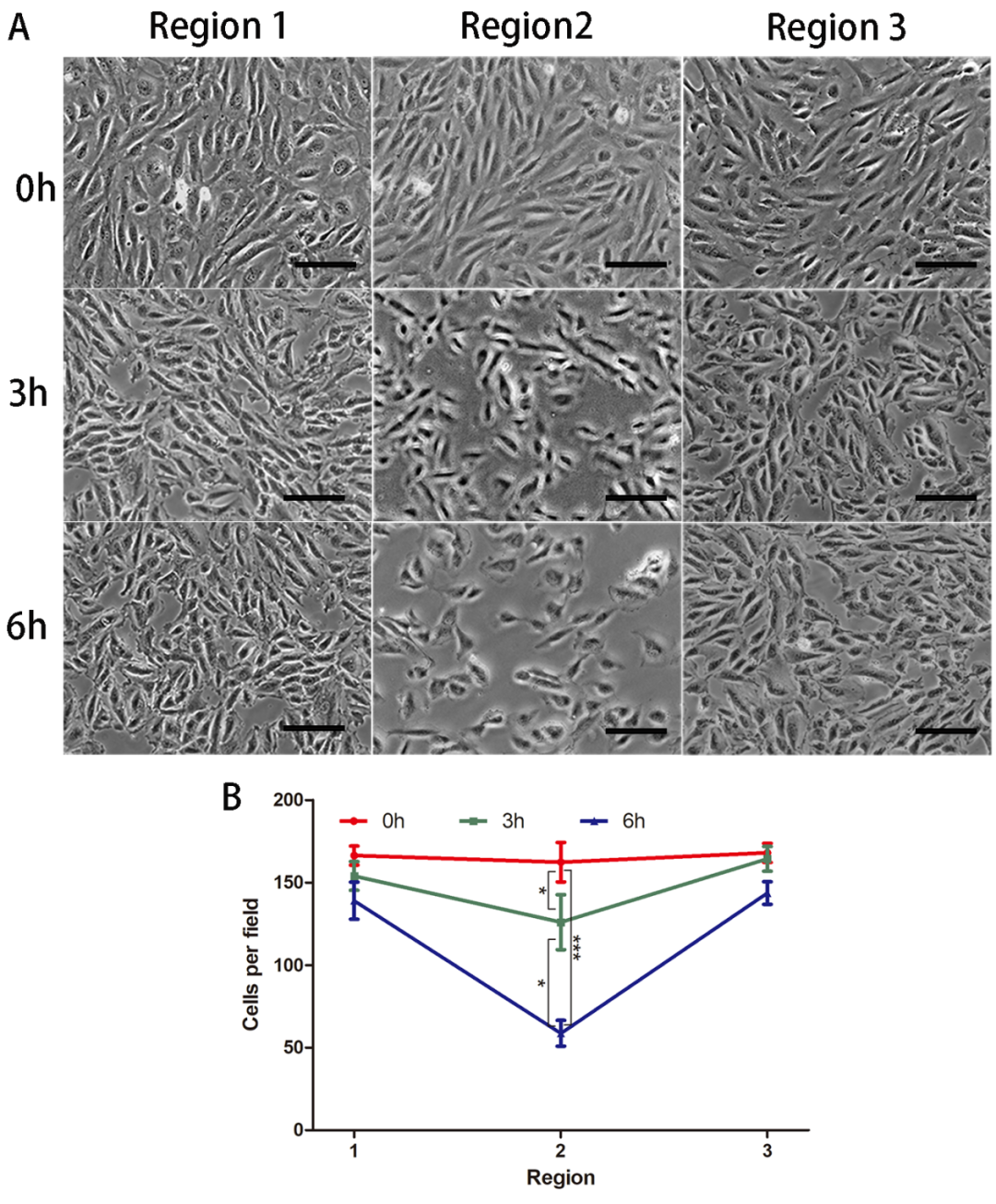
Changes in HUVECs Morphology
Under impinging flow, there were no significant changes in cell density in Regions 1 and 3, while there was a marked decrease in endothelial cell density in Region 2, with increased gaps between endothelial cells and diverse cell morphologies. Furthermore, as the duration of impinging flow increased, the cell density in Region 2 progressively decreased (Figure 2).
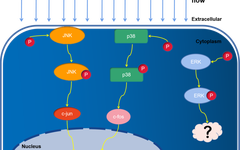
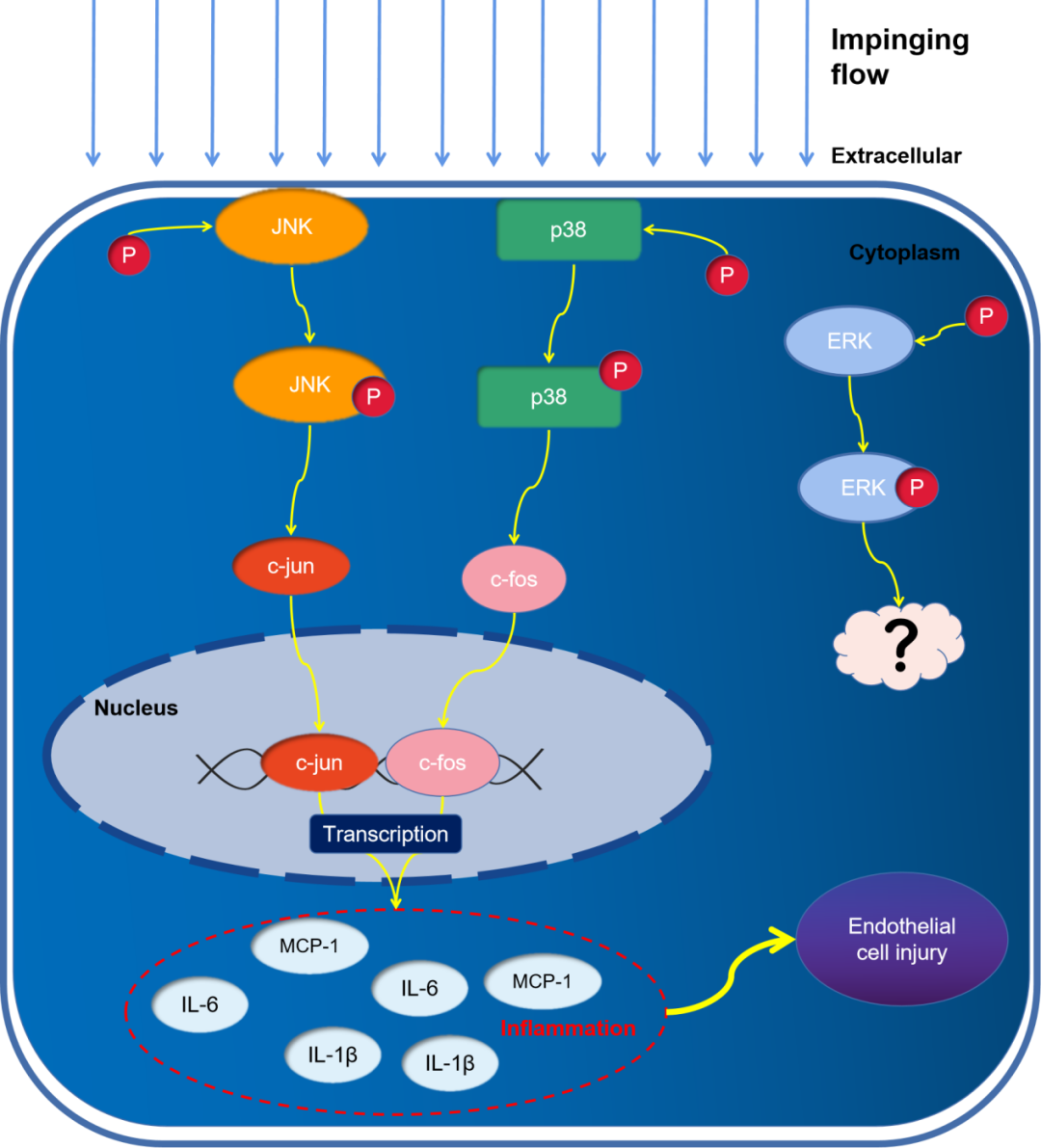
MAPK Activation and Expression of MCP-1 and Inflammatory Factors
HUVECs were exposed to impinging flow for 3 or 6 hours, and total protein was extracted from all three regions for WB analysis. Compared to the static control group, phosphorylation of ERK, JNK, and p38 increased over time. The expression levels of c-Fos, c-Jun, MCP-1, and inflammatory factors were significantly increased compared to the static control group (P < 0.05) (Figure 3).

Figure 3. Activation of Mitogen-Activated Protein Kinases (MAPK) and expression of MCP-1 and inflammatory factors. Under impinging flow, phosphorylation of ERK, JNK, and p38 increased over time (A-C), and expression levels of MCP-1, c-Jun, and c-Fos increased (D, E). mRNA expression of MCP-1, IL-1b, and IL-6 increased (F-H). Data are presented as mean ± standard error. *P<0.05, each experiment performed at least three times. *P<0.05, **P<0.01. ns, not significant.
JNK/c-Jun Pathway Regulates Expression of MCP-1 and Inflammatory Factors
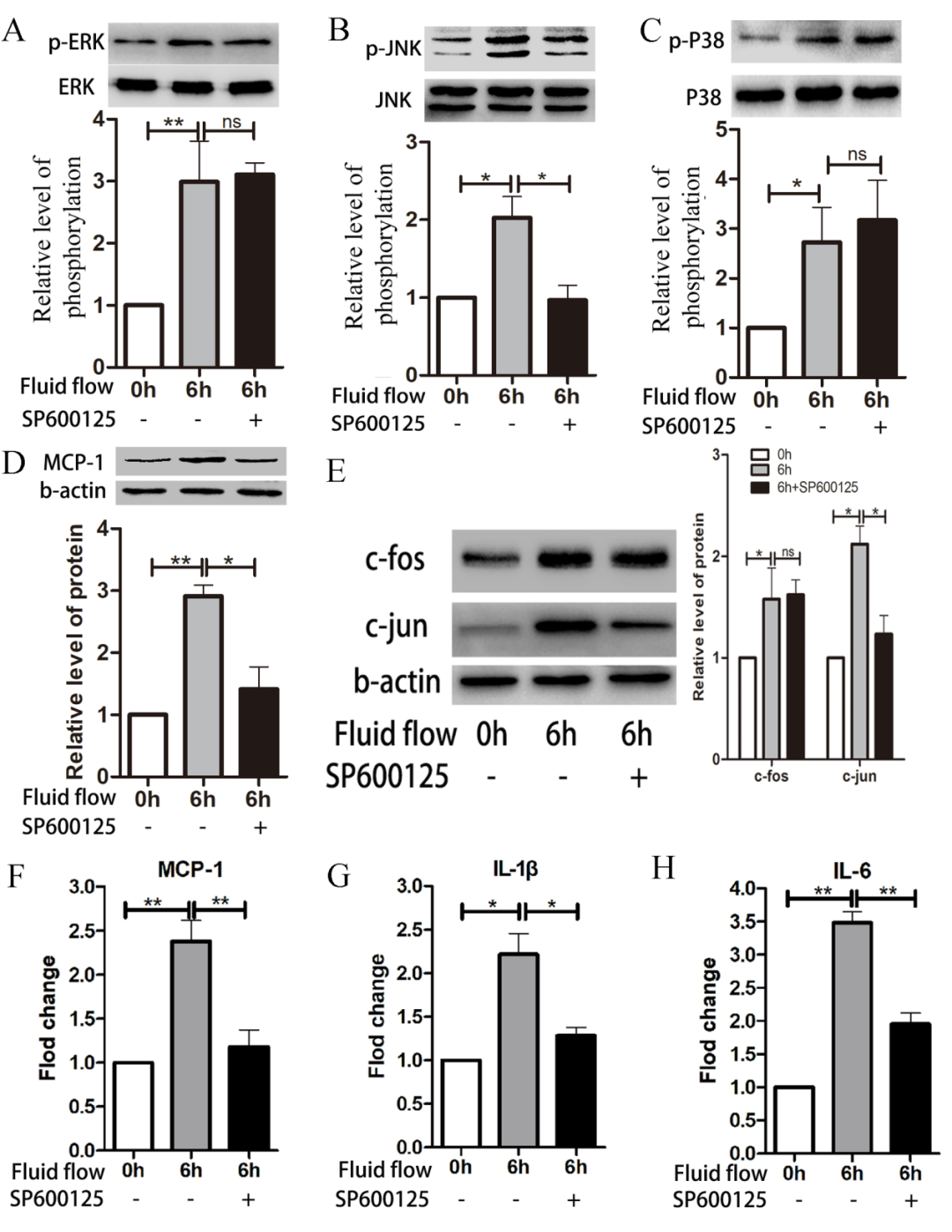
Figure 4. JNK/c-Jun pathway regulates expression of MCP-1 and inflammatory factors. HUVECs were pre-treated with the JNK inhibitor SP600125 (10μM) before exposure to impinging flow for 6 hours; phosphorylation of JNK decreased (B), while phosphorylation of ERK (A) and p38 (C) did not change. Expression levels of MCP-1 (D) and c-Jun (E) decreased, while expression of c-Fos (E) did not change. mRNA levels of MCP-1 (F), IL-1b (G), and IL-6 (H) decreased. Data are presented as mean ± standard error. Each experiment was performed at least three times. *P<0.05, **P<0.01. ns, not significant.
p38/c-Fos Pathway Regulates Expression of MCP-1 and Inflammatory Factors
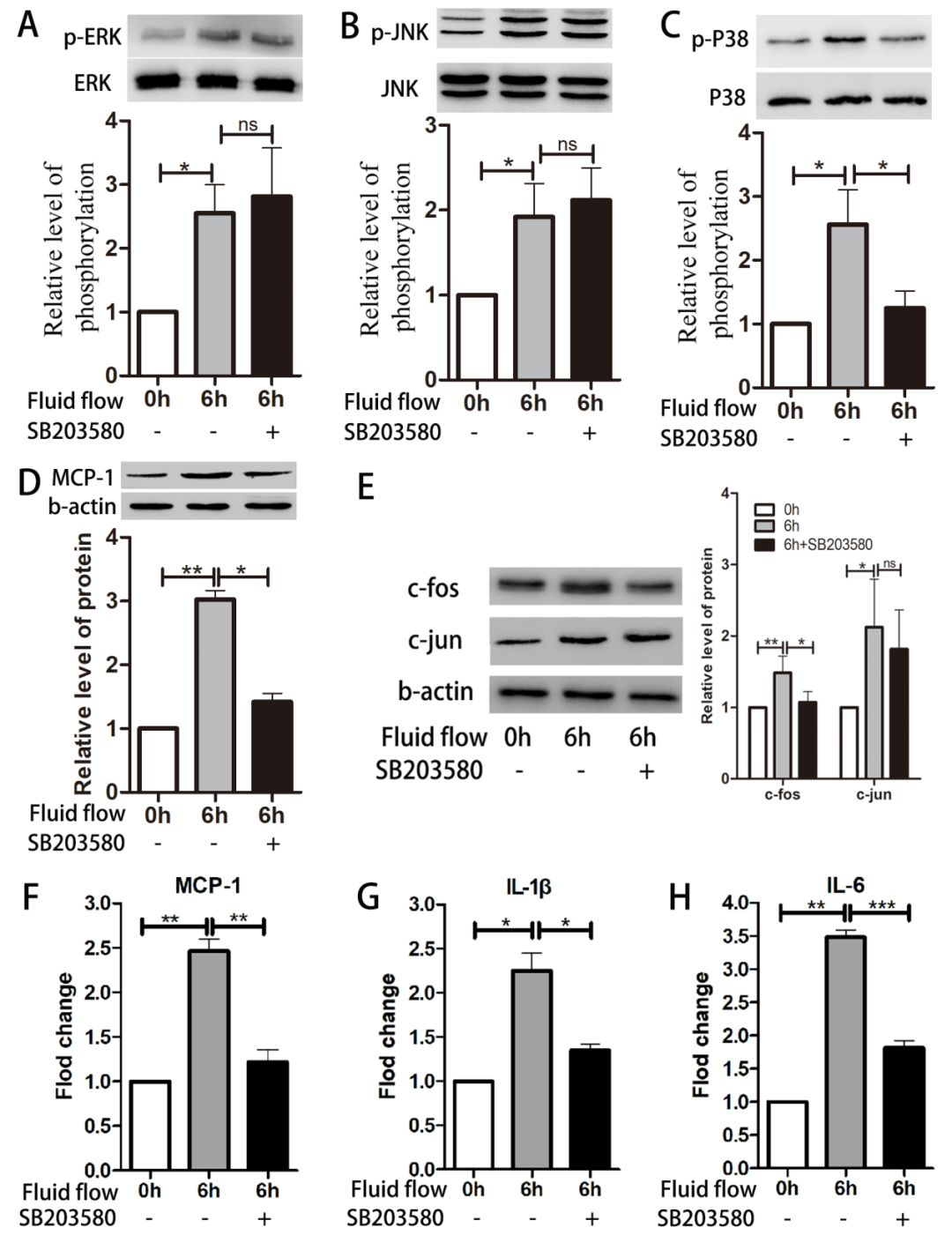
Figure 5. p38/c-Fos pathway regulates expression of MCP-1 and inflammatory factors. HUVECs were pre-treated with the p38 inhibitor SB203580 (10μM) before exposure to impinging flow for 6 hours; phosphorylation of p38 (C) decreased, while phosphorylation of ERK (A) and JNK (B) did not change. Expression levels of MCP-1 (D) and c-Fos (E) decreased, while expression of c-Jun (E) did not change. mRNA levels of MCP-1 (F), IL-1b (G), and IL-6 (H) decreased. Data are presented as mean ± standard error. Each experiment was performed at least three times. *P<0.05, **P<0.01. ns, not significant.
Inhibition of ERK Phosphorylation Does Not Significantly Affect Expression of MCP-1 and Inflammatory Factors
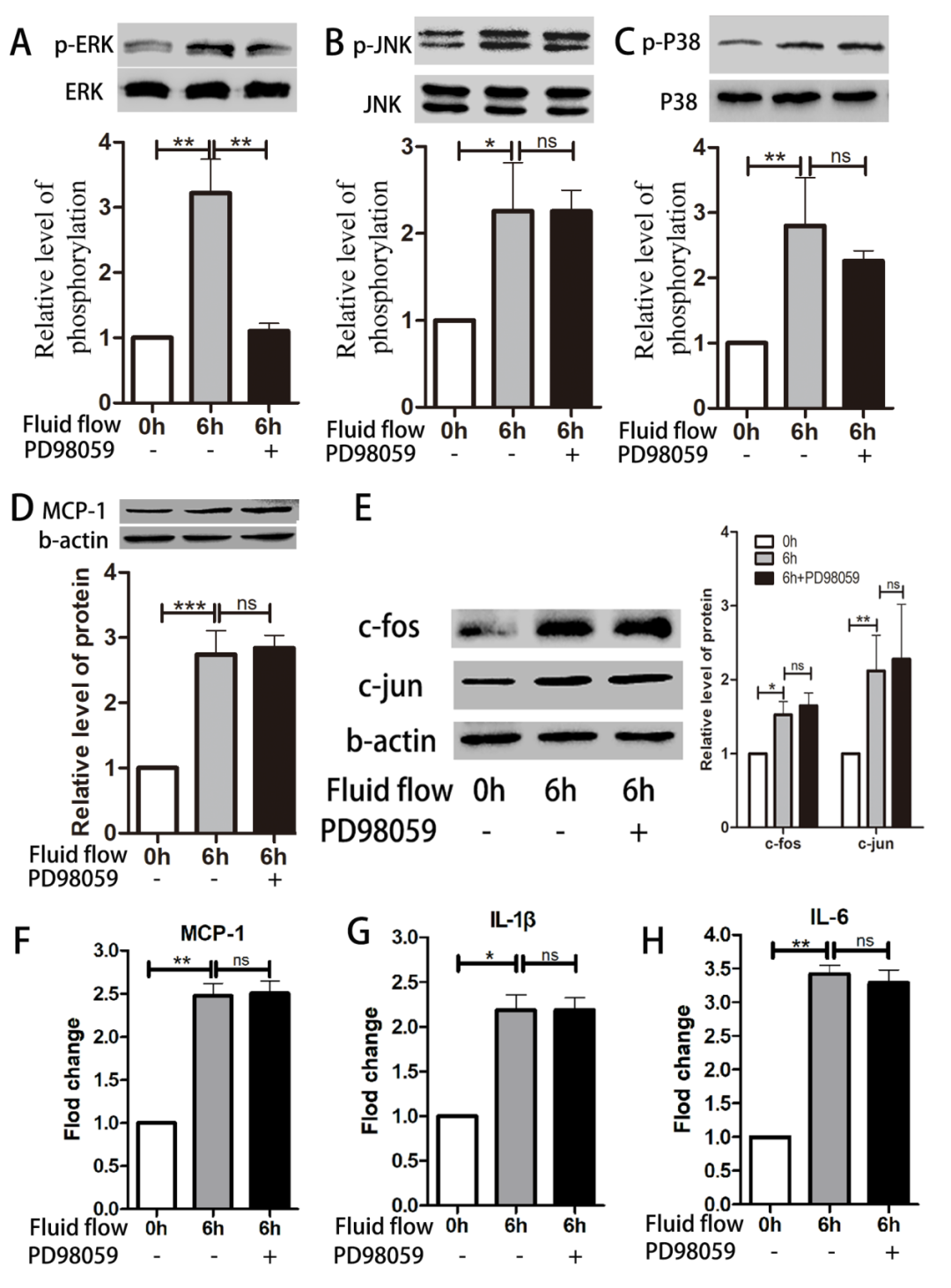
Figure 6. Inhibition of ERK phosphorylation does not significantly affect expression levels of MCP-1 and inflammatory factors. HUVECs were pre-treated with the ERK inhibitor PD98059 (10μM) before exposure to impinging flow for 6 hours; phosphorylation of ERK (A) decreased, while phosphorylation of JNK (B) and p38 (C) did not change. Expression levels of MCP-1 (D), c-Jun, and c-Fos (E) also did not change, similar to the mRNA levels of MCP-1 (F), IL-1b (G), and IL-6 (H). Data are presented as mean ± standard error. Each experiment was performed at least three times. *P<0.05, **P<0.01, ***P<0.001. ns, not significant.
Discussion
IA is a saccular arterial protrusion primarily caused by local thinning and dilation of the vascular wall. Rupture of IA is the leading cause of subarachnoid hemorrhage (SAH), resulting in high mortality and morbidity[48]. The mechanisms of IA formation and rupture have not been fully elucidated, but many studies link them to endothelial dysfunction, extracellular matrix remodeling, and vascular smooth muscle cell (VSMC) phenotypic changes that cause pathological alterations in the vascular system[4,19,20,29,30]. The lifecycle of IA has three distinct stages: formation, growth, and rupture. In each stage, blood flow plays a crucial role, converting mechanical stimuli into biological signals[45,49]. The fact that most lesions are located at arterial bifurcations or curves supports the importance of hemodynamics in the biology of IA[6,12]. The complex flow patterns determine the localization of EC dysfunction.[40,51].
ECs, forming a monolayer, are an important part of regulating vascular health; the endothelial layer is regularly arranged throughout the vascular network, forming a molecular barrier that is crucial for various physiological functions[49]. Under normal physiological conditions, vascular ECs become spindle-shaped, aligning with the flow of blood to regulate vascular tension, cell adhesion, and proliferation of smooth muscle cells, thereby maintaining vascular homeostasis[7]. Abnormal hemodynamic conditions trigger EC dysfunction and injury; subsequently, ECs secrete matrix metalloproteinases (such as MMP2 and MMP), damaging the extracellular matrix[41,47,54]. Flow-mediated endothelial dysfunction, followed by VSMC phenotypic transition from contractile to synthetic (thereby enhancing the inflammatory response), is a key step in the pathophysiological development of IA[49]. The synthetic phenotype promotes inflammation in many vascular diseases by creating an environment conducive to inflammation and remodeling, which in turn maintains and expands endothelial dysfunction and recruitment of immune cells[38,46]. Thus, endothelial dysfunction is a core pathogenesis of IA and may indeed be the initial step in IA formation. We simulated hemodynamics at arterial bifurcations using a modified T-shaped flow chamber to explore how impinging flow affects the behavior of ECs[60]. Our results indicate that impinging flow disrupts cell-cell junctions in HUVECs, activates intracellular inflammatory responses, and subsequently triggers endothelial dysfunction.
The chemokine MCP-1 has a powerful effect on both monocytes and macrophages. Significant macrophage infiltration has been observed in both ruptured and unruptured IAs, and inhibiting macrophages can significantly reduce IA formation[23]. Hemodynamics associated with high WSS activate pro-inflammatory signals in vascular ECs, leading to the accumulation of macrophages at sites exposed to high WSS and triggering inflammatory responses[14,21]. Previous studies have found high expression of MCP-1 in IA walls, and knockout of MCP-1 significantly reduced macrophage infiltration, confirming its important regulatory role in IA formation[2]. In this study, we simulated fluid flow in vitro and found that MCP-1 expression in HUVECs significantly increased under impinging flow; this provides evidence for MCP-1’s involvement in the early stages of endothelial dysfunction.
The MAPK signaling pathway plays a crucial role in signal transduction from the cell surface to the interior; it is also involved in cell proliferation, differentiation, migration, and apoptosis, as well as stress and inflammation. The MAPK pathway transmits extracellular signals to cells through receptors (including G protein-coupled receptors), protein kinases, and transcription factors[18]. We found that phosphorylation of ERK, JNK, and p38 was activated by impinging flow. We confirmed using specific inhibitors that JNK and p38 regulate the expression of MCP-1 and inflammatory factors via c-Jun and c-Fos, respectively. Although ERK plays a limited role in the production of MCP-1 and inflammatory factors, phosphorylation/activation of ERK triggers downstream signaling pathways involved in regulating cell growth, proliferation, differentiation, and migration, leading to widespread cellular changes[8]. After comparing differentially expressed genes in IA and normal superficial temporal artery tissues, it was found that the ERK pathway may play an important role in the formation and development of IA. Therefore, further research is needed to determine how ERK may be involved in regulating endothelial dysfunction (through other biological pathways)[58].
Impinging flow regulates the inflammatory response of HUVECs by activating JNK and p38, which may participate in the formation and development of IA by modulating endothelial dysfunction. During IA formation, JNK and p38 regulate the phenotypic transition of VSMCs[55]. Compared to unruptured aneurysms, phosphorylation of JNK and p38 is enhanced in ruptured IA tissues and positively correlates with IA size[32,33]. Increased levels of ANXA3 and enhanced JNK signaling activity can be observed in IA; silencing ANXA3 can inhibit the phenotypic transition of VSMCs in IA by suppressing phosphorylation of the JNK signaling pathway[52]. MiR-21 promotes the production of inflammatory-related factors through the JNK signaling pathway, participating in the formation and rupture of IA[13]. In a rabbit IA model, upregulation of KLF2 increased phosphorylation of p38 and regulated IA progression[53]. Although many studies have found that JNK and p38 are involved in the occurrence and development of IA, the cellular processes regulating their activity are very complex. It remains unclear whether impinging flow participates in IA by modulating MCP-1 and inflammation (mediated by JNK and p38).
Our work has certain limitations. First, the cells from the three regions affected by impinging flow were collected together, and we did not analyze the cells from different regions separately. Considering the limited size of the slides and the limited number of cultured cells, the number of cells may be too small for subsequent analyses due to the reduction caused by impinging flow. If the cells were collected by region, their numbers might be too few for further analysis. However, given the advancements in sequencing technology, particularly single-cell sequencing, we may analyze cells from different regions in the future to strengthen our findings on the effects of impinging flow on HUVECs and related signaling pathways in vitro. Furthermore, this study lacks validation from animal IA models. Third, vascular endothelial cells constitute only part of the arterial wall, and other components of the artery still need further investigation to determine their impact on IA.
Conclusion
Discussion
Under impinging flow, MCP-1 and inflammatory factors are regulated via the JNK/c-Jun/p38/c-Fos pathway, participating in endothelial dysfunction. However, further in vivo experimental validation is necessary.
References
1. Ali M, Starke R, Jabbour P, et al. TNF-α induces phenotypic modulation in cerebral vascular smooth muscle cells: implications for cerebral aneurysm pathology. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2013;33(10):1564-1573. https://doi.org/10.1038/jcbfm.2013.109.
2. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Egashira K, Hashimoto N. Impact of monocyte chemoattractant protein-1 deficiency on cerebral aneurysm formation. Stroke. 2009;40(3):942-951. https://doi.org/10.1161/STROKEAHA.108.532556.
3. Bao R, Hou J, Li Y, et al. Adenosine promotes Foxp3 expression in Treg cells in sepsis model by activating JNK/AP-1 pathway. Am J Transl Res. 2016;8(5):2284-2292.
4. Bor AS, Tiel GA, TerBrugge KG, et al. Clinical, radiological, and flow-related risk factors for growth of untreated, unruptured intracranial aneurysms. Stroke. 2015;46(1):42-48. https://doi.org/10.1161/STROKEAHA.114.005963.
5. Braicu C, Buse M, Busuioc C, et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers. 2019;11(10). https://doi.org/10.3390/cancers11101618.
6. Brisman JL, Song JK, Newell DW. Cerebral aneurysms. N Engl J Med. 2006;355(9):928-939. https://doi.org/10.1056/NEJMra052760.
7. Brooks A, Lelkes P, Rubanyi G. Gene expression profiling of vascular endothelial cells exposed to fluid mechanical forces: relevance for focal susceptibility to atherosclerosis. Endothelium : journal of endothelial cell research. 2004;11(1):45-57. https://doi.org/10.1080/10623320490432470.
8. Cao Z, Liao Q, Su M, Huang K, Jin J, Cao D. AKT and ERK dual inhibitors: The way forward? Cancer Lett. 2019;459:30-40. https://doi.org/10.1016/j.canlet.2019.05.025.
9. Castro MA, Putman CM, Sheridan MJ, Cebral JR. Hemodynamic patterns of anterior communicating artery aneurysms: a possible association with rupture. AJNR Am J Neuroradiol. 2009;30(2):297-302. https://doi.org/10.3174/ajnr.A1323.
10. Cebral J, Mut F, Weir J, Putman C. Quantitative characterization of the hemodynamic environment in ruptured and unruptured brain aneurysms. AJNR. American journal of neuroradiology. 2011;32(1):145-151. https://doi.org/10.3174/ajnr.A2419.
11. Cebral JR, Mut F, Weir J, Putman CM. Association of hemodynamic characteristics and cerebral aneurysm rupture. AJNR Am J Neuroradiol. 2011;32(2):264-270. https://doi.org/10.3174/ajnr.A2274.
12. Chen S, Bai B, Lv N, Cheng Y, Ji B. Hemodynamic analysis and implantation strategies of delayed intracranial aneurysm rupture after flow diverter treatment. Ann Transl Med. 2021;9(23):1735. https://doi.org/10.21037/atm-21-5939.
13. Chen Z, Song S, Zhu J, Lai X. Regulatory mechanism of MiR-21 in formation and rupture of intracranial aneurysm through JNK signaling pathway-mediated inflammatory response. Int J Clin Exp Patho. 2020.
14. Chu C, Xu G, Li X, et al. Sustained expression of MCP-1 induced low wall shear stress loading in conjunction with turbulent flow on endothelial cells of intracranial aneurysm. J Cell Mol Med. 2021;25(1):110-119. https://doi.org/10.1111/jcmm.15868.
15. Dabagh M, Nair P, Gounley J, Frakes D, Gonzalez LF, Randles A. Hemodynamic and morphological characteristics of a growing cerebral aneurysm. Neurosurg Focus. 2019;47(1):E13. https://doi.org/10.3171/2019.4.FOCUS19195.
16. Davies PF. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat Clin Pract Cardiovasc Med. 2009;6(1):16-26. https://doi.org/10.1038/ncpcardio1397.
17. Etminan N, Rinkel GJ. Unruptured intracranial aneurysms: development, rupture and preventive management. Nat Rev Neurol. 2016;12(12):699-713. https://doi.org/10.1038/nrneurol.2016.150.
18. Fang JY, Richardson BC. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005;6(5):322-327. https://doi.org/10.1016/S1470-2045(05)70168-6.
19. Fennell VS, Kalani MY, Atwal G, Martirosyan NL, Spetzler RF. Biology of Saccular Cerebral Aneurysms: A Review of Current Understanding and Future Directions. Front Surg. 2016;3:43. https://doi.org/10.3389/fsurg.2016.00043.
20. Frosen J. Smooth muscle cells and the formation, degeneration, and rupture of saccular intracranial aneurysm wall–a review of current pathophysiological knowledge. Transl Stroke Res. 2014;5(3):347-356. https://doi.org/10.1007/s12975-014-0340-3.
21. Frösen J, Cebral J, Robertson A, Aoki T. Flow-induced, inflammation-mediated arterial wall remodeling in the formation and progression of intracranial aneurysms. Neurosurg Focus. 2019;47(1):E21. https://doi.org/10.3171/2019.5.FOCUS19234.
22. Frosen J, Tulamo R, Paetau A, et al. Saccular intracranial aneurysm: pathology and mechanisms. Acta Neuropathol. 2012;123(6):773-786. https://doi.org/10.1007/s00401-011-0939-3.
23. Fukuda M, Aoki T. Molecular basis for intracranial aneurysm formation. Acta Neurochir Suppl. 2015;120:13-15. https://doi.org/10.1007/978-3-319-04981-6_2.
24. Hao Z, Li Y, Yu N, et al. Analysis of differentially expressed circular RNAs in endothelial cells under impinging flow. Mol Cell Probes. 2020;51:101539. https://doi.org/10.1016/j.mcp.2020.101539.
25. Hasan D, Chalouhi N, Jabbour P, Hashimoto T. Macrophage imbalance (M1 vs. M2) and upregulation of mast cells in wall of ruptured human cerebral aneurysms: preliminary results. J Neuroinflamm. 2012;9:222. https://doi.org/10.1186/1742-2094-9-222.
26. Jamous M, Nagahiro S, Kitazato K, et al. Endothelial injury and inflammatory response induced by hemodynamic changes preceding intracranial aneurysm formation: experimental study in rats. J Neurosurg. 2007;107(2):405-411. https://doi.org/10.3171/JNS-07/08/0405.
27. Kanematsu Y, Kanematsu M, Kurihara C, et al. Critical roles of macrophages in the formation of intracranial aneurysm. Stroke. 2011;42(1):173-178. https://doi.org/10.1161/STROKEAHA.110.590976.
28. Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802(4):396-405. https://doi.org/10.1016/j.bbadis.2009.12.009.
29. Kleinloog R, de Mul N, Verweij BH, Post JA, Rinkel G, Ruigrok YM. Risk Factors for Intracranial Aneurysm Rupture: A Systematic Review. Neurosurgery. 2018;82(4):431-440. https://doi.org/10.1093/neuros/nyx238.
30. Krings T, Mandell DM, Kiehl TR, et al. Intracranial aneurysms: from vessel wall pathology to therapeutic approach. Nat Rev Neurol. 2011;7(10):547-559. https://doi.org/10.1038/nrneurol.2011.136.
31. Kwon OK. Headache and Aneurysm. Neuroimaging Clin N Am. 2019;29(2):255-260. https://doi.org/10.1016/j.nic.2019.01.004.
32. Laaksamo E, Ramachandran M, Frösen J, et al. Intracellular signaling pathways and size, shape, and rupture history of human intracranial aneurysms. Neurosurgery. 2012.
33. Laaksamo E, Tulamo R, Baumann M, et al. Involvement of mitogen-activated protein kinase signaling in growth and rupture of human intracranial aneurysms. Stroke. 2008.
34. Lauric A, Hippelheuser JE, Malek AM. Induction of aneurysmogenic high positive wall shear stress gradient by wide angle at cerebral bifurcations, independent of flow rate. J Neurosurg. 2018;131(2):442-452. https://doi.org/10.3171/2018.3.JNS173128.
35. Lee W, Mitchell P, Tjian R. Purified transcription factor AP-1 interacts with TPA-inducible enhancer elements. Cell. 1987;49(6):741-752. https://doi.org/10.1016/0092-8674(87)90612-x.
36. Meng H, Tutino VM, Xiang J, Siddiqui A. High WSS or low WSS? Complex interactions of hemodynamics with intracranial aneurysm initiation, growth, and rupture: toward a unifying hypothesis. AJNR Am J Neuroradiol. 2014;35(7):1254-1262. https://doi.org/10.3174/ajnr.A3558.
37. Nixon A, Gunel M, Sumpio B. The critical role of hemodynamics in the development of cerebral vascular disease. J Neurosurg. 2010;112(6):1240-1253. https://doi.org/10.3171/2009.10.JNS09759.
38. Orr AW, Hastings NE, Blackman BR, Wamhoff BR. Complex regulation and function of the inflammatory smooth muscle cell phenotype in atherosclerosis. J Vasc Res. 2010;47(2):168-180. https://doi.org/10.1159/000250095.
39. Park S, Won S, Song J, et al. EPO gene expression promotes proliferation, migration and invasion via the p38MAPK/AP-1/MMP-9 pathway by p21WAF1 expression in vascular smooth muscle cells. Cell Signal. 2015;27(3):470-478. https://doi.org/10.1016/j.cellsig.2014.12.001.
40. Penn DL, Komotar RJ, Sander CE. Hemodynamic mechanisms underlying cerebral aneurysm pathogenesis. J Clin Neurosci. 2011;18(11):1435-1438. https://doi.org/10.1016/j.jocn.2011.05.001.
41. Ramella M, Boccafoschi F, Bellofatto K, et al. Endothelial MMP-9 drives the inflammatory response in abdominal aortic aneurysm (AAA). Am J Transl Res. 2017;9(12):5485-5495.
42. Shao L, Qin X, Liu J, Jian Z, Xiong X, Liu R. Macrophage Polarization in Cerebral Aneurysm: Perspectives and Potential Targets. J Immunol Res. 2017;2017:8160589. https://doi.org/10.1155/2017/8160589.
43. Sheinberg DL, McCarthy DJ, Elwardany O, et al. Endothelial dysfunction in cerebral aneurysms. Neurosurg Focus. 2019;47(1):E3. https://doi.org/10.3171/2019.4.FOCUS19221.
44. Shimizu K, Kushamae M, Mizutani T, Aoki T. Intracranial Aneurysm as a Macrophage-mediated Inflammatory Disease. Neurol Med Chir (Tokyo). 2019;59(4):126-132. https://doi.org/10.2176/nmc.st.2018-0326.
45. Shojima M, Oshima M, Takagi K, et al. Magnitude and role of wall shear stress on cerebral aneurysm: computational fluid dynamic study of 20 middle cerebral artery aneurysms. Stroke. 2004;35(11):2500-2505. https://doi.org/10.1161/01.STR.0000144648.89172.0f.
46. Sibon I, Mercier N, Darret D, Lacolley P, Lamaziere JM. Association between semicarbazide-sensitive amine oxidase, a regulator of the glucose transporter, and elastic lamellae thinning during experimental cerebral aneurysm development: laboratory investigation. J Neurosurg. 2008;108(3):558-566. https://doi.org/10.3171/JNS/2008/108/3/0558.
47. Staarmann B, Smith M, Prestigiacomo CJ. Shear stress and aneurysms: a review. Neurosurg Focus. 2019;47(1):E2. https://doi.org/10.3171/2019.4.FOCUS19225.
48. Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G. European Stroke Organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis. 2013;35(2):93-112. https://doi.org/10.1159/000346087.
49. Turjman AS, Turjman F, Edelman ER. Role of fluid dynamics and inflammation in intracranial aneurysm formation. Circulation. 2014;129(3):373-382. https://doi.org/10.1161/CIRCULATIONAHA.113.001444.
50. Vlak M, Algra A, Brandenburg R, Rinkel G. Prevalence of unruptured intracranial aneurysms, with emphasis on sex, age, comorbidity, country, and time period: a systematic review and meta-analysis. The Lancet. Neurology. 2011;10(7):626-636. https://doi.org/10.1016/S1474-4422(11)70109-0.
51. Wang H, Balzani D, Vedula V, Uhlmann K, Varnik F. On the Potential Self-Amplification of Aneurysms Due to Tissue Degradation and Blood Flow Revealed From FSI Simulations. Front Physiol. 2021;12:785780. https://doi.org/10.3389/fphys.2021.785780.
52. Wang Y, Wang C, Yang Q, Cheng Y. ANXA3 Silencing Ameliorates Intracranial Aneurysm via Inhibition of the JNK Signaling Pathway. Molecular therapy. Nucleic acids. 2019.
53. Wu X, Zhang J, Yang P, Huang Q, Liu J. Regulation of Kruppel-like factor 2 (KLF2) in the pathogenesis of intracranial aneurysm induced by hemodynamics. Am J Transl Res. 2017.
54. Xu Y, Zhang B, Chen Y, et al. Simvastatin increases circulating endothelial progenitor cells and inhibits the formation of intracranial aneurysms in rats with diet-induced hyperhomocysteinemia. Neurosci Lett. 2021;760:136072. https://doi.org/10.1016/j.neulet.2021.136072.
55. Yan Y, Xiong J, Xu F, et al. SDF-1α/CXCR4 Pathway Mediates Hemodynamics-Induced Formation of Intracranial Aneurysm by Modulating the Phenotypic Transformation of Vascular Smooth Muscle Cells. Transl Stroke Res. 2021.
56. Yang H, Jiang H, Ni W, et al. Treatment Strategy for Unruptured Intracranial Aneurysm in Elderly Patients: Coiling, Clipping, or Conservative? Cell Transplant. 2019;28(6):767-774. https://doi.org/10.1177/0963689718823517.
57. Ye N, Ding Y, Wild C, Shen Q, Zhou J. Small molecule inhibitors targeting activator protein 1 (AP-1). J Med Chem. 2014;57(16):6930-6948. https://doi.org/10.1021/jm5004733.
58. Yu L, Wang J, Wang S, et al. DNA Methylation Regulates Gene Expression in Intracranial Aneurysms. World Neurosurg. 2017;105:28-36. https://doi.org/10.1016/j.wneu.2017.04.064.
59. Zhang HF, Zhao MG, Liang GB, Song ZQ, Li ZQ. Expression of pro-inflammatory cytokines and the risk of intracranial aneurysm. Inflammation. 2013;36(6):1195-1200. https://doi.org/10.1007/s10753-013-9655-6.
60. Zhao JL, Jia L, Wang XB, Zhang LL, Li MH. Effects of adjustable impinging flow on the vascular endothelial cell layer in a modified T chamber. Int J Clin Exp Med. 2017;10(3):5068-5074.
61. Zhao JL, Xiao ZP, Yu NZ, Jiang JW, Li MH. Knockdown of P120 catenin aggravates endothelial injury under an impinging flow by inducing breakdown of adherens junctions. Mol Med Rep. 2019;19(1):541-548. https://doi.org/10.3892/mmr.2018.9657.
62. Zhao YY, Huang SX, Hao Z, Zhu HX, Xing ZL, Li MH. Fluid Shear Stress Induces Endothelial Cell Injury via Protein Kinase C Alpha-Mediated Repression of p120-Catenin and Vascular Endothelial Cadherin In Vitro. World Neurosurg. 2020;136:e469-e475. https://doi.org/10.1016/j.wneu.2020.01.028.
63. Zhu H, Tan J, Zhao Y, Wang Z, Wu Z, Li M. Potential Role of the Chemotaxis System in Formation and Progression of Intracranial Aneurysms Through Weighted Gene Co-Expression Network Analysis. Int J Gen Med. 2022;15:2217-2231. https://doi.org/10.2147/IJGM.S347420.
64. Zimny M, Kawlewska E, Hebda A, Wolański W, Ładziński P, Kaspera W. Wall shear stress gradient is independently associated with middle cerebral artery aneurysm development: a case-control CFD patient-specific study based on 77 patients. Bmc Neurol. 2021;21(1):281. https://doi.org/10.1186/s12883-021-02251-3.
Corresponding Author Profile

Professor Li Meihua
First Affiliated Hospital of Nanchang University
-
Director of Neurosurgery, Chief Physician, Associate Professor, Doctoral Supervisor
-
Outstanding Young and Middle-aged Expert in Jiangxi Province Health System, Jiangxi Province Talent Program, Jiangxi Province Backbone Teacher and Young and Middle-aged Discipline Leader
-
In 2016, awarded the second prize of Science and Technology Progress Award for “Clinical Application of Lateral Cranial Base Approach for Complex Cranial Base Tumors” (first contributor) and in 2020, awarded the first prize of Jiangxi Province Science and Technology Progress Award for “Construction and Application of New System for Diagnosis and Treatment of Craniopharyngioma” (second contributor)

Scan the QR code to visitProfessor Li Meihua’s Academic Homepage
View more exciting content

Brain Medical Exchange App (formerly Neurosurgery News/ Neurointervention News App) has launched a new consultation room for patients, answering patient inquiries in fragmented time. Click “Read the original text” to experience it first!