Authors: Zhao Xiaoyu, Jiang Nan, Liu Chunyan, Fu Rong
Author Affiliation: Department of Hematology, Tianjin Medical University General Hospital
Corresponding Author: Fu Rong, [email protected]
Funding: National Natural Science Foundation (81870101, 81970115)
Citation Format: Zhao Xiaoyu, Jiang Nan, Liu Chunyan, et al. Case Report: Hemophagocytic Lymphohistiocytosis with Rag1 Gene Mutation [DB/OL]. China Clinical Case Achievement Database, 2022 (2022-11-09). http://journal.yiigle.com/LinkIn.do?linkin_type=cma&DOI=10.3760/cma.j.cmcr.2022.e06819.
Abstract
Case Summary One month prior to admission, the patient developed abdominal distension and pain after self-medicating with antihistamines for urticaria, accompanied by poor appetite and vomiting, with vomit consisting of gastric contents and constipation, defecating approximately once a week with clay-colored stools, intermittent fever, cough, and expectorating white sputum. Treatment with cephalosporins, Asmei, and Lianhua Qingwen capsules resulted in slight improvement of the cough. Subsequently, the patient developed yellowing of the sclera and skin, accompanied by tea-colored urine, with normal urine output. The patient was then referred to Tianjin Third Central Hospital, where persistent fever was noted, with a maximum temperature of 39°C. Treatment with ertapenem, piperacillin, and thiamphenicol for infection, along with dexamethasone, was initiated. During hospitalization, the patient experienced seizures, with upward eye movement, foaming at the mouth, flexion of limbs, and altered consciousness. The patient had lost 5 kg since the onset of illness.
Symptoms and Signs Abdominal distension, abdominal pain, with severe jaundice of the skin and mucous membranes.
Diagnostic Methods The patient exhibited significantly elevated soluble CD25 levels, decreased NK cell activity; bone marrow examination showed significant hemophagocytosis; concurrently, a Rag1 gene mutation unrelated to hemophagocytic lymphohistiocytosis was identified.
Treatment Methods Primarily symptomatic treatment including anti-infection, antiviral, cough suppression, antiemetic, and liver protection. Treatment included injectable tigecycline, injectable methylprednisolone sodium succinate, piperacillin-thiabendazole, injectable ganciclovir for antiviral treatment, anti-infection, magnesium isoglycyrrhizinate injection, ademetionine for liver protection, enteric-coated rabeprazole for gastric protection, and adequate potassium supplementation with potassium chloride sustained-release tablets. Rivaroxaban tablets were administered to stabilize the cervical wall thrombus. Micafungin was given for prophylactic antifungal treatment. Oral furosemide and spironolactone were used for diuresis. Injectable doxofylline, ipratropium bromide nebulization solution, and inhaled budesonide suspension were used for cough suppression and asthma relief. Metoclopramide hydrochloride was administered for antiemetic treatment, and recombinant human erythropoietin was used to improve blood parameters.
Clinical Outcome After treatment, the patient recovered from the disease and has been followed up to date, with all medications discontinued and complete recovery.
Target Audience Hematology professionals
Hemophagocytic lymphohistiocytosis (HLH) is a rare, fatal syndrome characterized by a strong immune activation. The most typical findings of HLH are fever, hepatosplenomegaly, and cytopenias. Other common findings include hypertriglyceridemia, coagulopathy with hypofibrinogenemia, liver dysfunction, elevated ferritin and serum transaminase levels, and neurological symptoms associated with cerebrospinal fluid abnormalities and neutrophilia. In summary, HLH is a pathological immune activation syndrome characterized by extreme inflammatory clinical signs and symptoms.
1. Susceptibility Immune Deficiencies:
1) Decreased or absent NK cell function;
2) Cytotoxic gene defects;
3) Family history of HLH;
4) Previous episodes of HLH or unexplained cytopenias;
5) Markers of cytotoxic impairment: decreased expression of perforin, SAP, XIAP, or mobilization of CD107a.
2. Significant Immune Activation
1) Fever;
2) Splenomegaly/hepatomegaly;
3) Elevated ferritin (>3000 ng/mL);
4) Elevated sCD25;
5) Elevated sCD163.
3. Abnormal Immune Pathology
1) Cytopenias;
2) Decreased fibrinogen or increased triglycerides;
3) Hemophagocytosis;
4) Hepatitis;
5) Central nervous system involvement;
4. Associated Gene Phenotypes
Clinical Data
1. General Information
The patient, a 17-year-old female, was admitted with the primary complaint of “fever, abdominal distension, and jaundice for over one month”.
History of Present Illness: One month prior to admission, the patient developed abdominal distension and pain after self-medicating with antihistamines for urticaria, accompanied by poor appetite and vomiting, with vomit consisting of gastric contents and constipation, defecating approximately once a week with clay-colored stools, intermittent fever, cough, and expectorating white sputum. Treatment with cephalosporins, Asmei, and Lianhua Qingwen capsules resulted in slight improvement of the cough. Subsequently, the patient developed yellowing of the sclera and skin, accompanied by tea-colored urine, with normal urine output. The patient was then referred to Tianjin Third Central Hospital, where persistent fever was noted, with a maximum temperature of 39°C. Treatment with ertapenem, piperacillin, and thiamphenicol for infection, along with dexamethasone, was initiated. During hospitalization, the patient experienced seizures, with upward eye movement, foaming at the mouth, flexion of limbs, and altered consciousness. The patient had lost 5 kg since the onset of illness.
Past Medical History: The patient was previously healthy. Personal History: No history of drug allergies. No smoking or alcohol use. Family History: The father was found to have a heterozygous Rag1 gene mutation upon examination. Admission Physical Examination: Severe jaundice of the skin and mucous membranes, no other abnormalities noted.
2. Examination
Laboratory tests and examinations after admission: Complete blood count: WBC 0.32×109/L, RBC 2.51×1012/L, hemoglobin 70 g/L, hematocrit 20.7%, platelets 68×109/L, neutrophil percentage 12.9%, monocyte percentage 2.7%, lymphocyte percentage 80.3%, eosinophil percentage 0.3%, basophil percentage 0.4%, absolute neutrophil count 0.04×107/L, absolute monocyte count 0.01×109/L, absolute eosinophil count 0.02×109/L, absolute basophil count 0.05×109/L. There was evidence of trilineage cytopenias; soluble CD25 level: 34445 (N<6400) significantly elevated; NK cell activity 5.24%, indicating decreased NK cell function or number; ADAMTS13 enzyme activity 284.88%, inhibitor antibody testing negative; bone marrow examination (Figure 1): decreased granulocyte proportion, early and immature granulocytes easily seen, significant nuclear-cytoplasmic developmental imbalance. Increased erythroid proportion, early and immature erythrocytes easily seen, predominantly mature erythrocytes, with some binucleated and trinucleated cells. Mature erythrocytes of varying sizes. Increased proportion of mature lymphocytes. Platelets scattered. Phagocytic cells easily seen, with phagocytic cells and platelets consistent with the bone marrow findings of hemophagocytic syndrome.
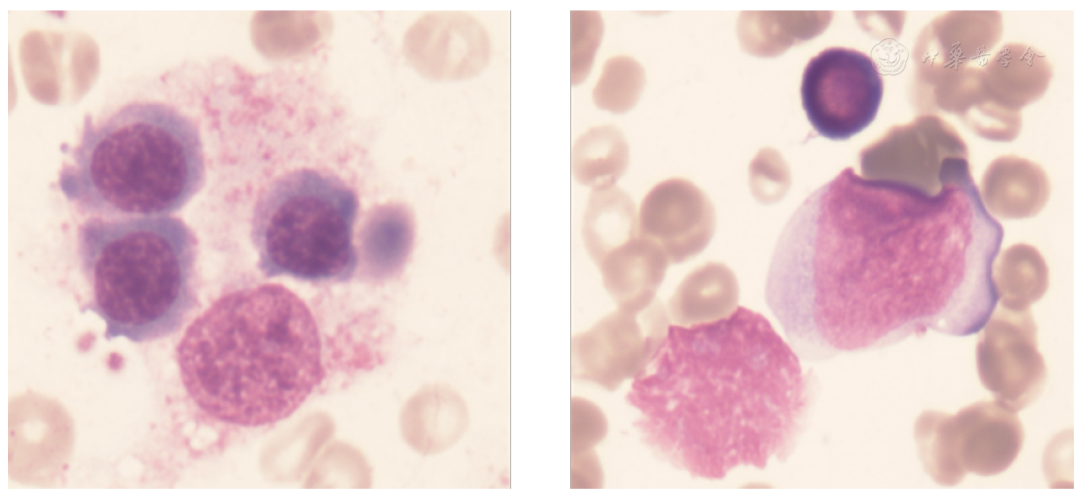
▲Figure 1 Bone marrow examination of the patient shows significant hemophagocytosis.
Gene testing report related to hemophagocytic syndrome: detection of Rag1 gene mutation, no pathogenic or suspected pathogenic single nucleotide variations detected, nor small fragment insertions or deletions and copy number variations. Detected heterozygous variation in Rag1, chromosome location chr11: 36597564, transcript number/exon NM_000448.3 (Exon 2), nucleotide change c.2710G>C, amino acid change p.E904Q, with the father being a heterozygous carrier of the same gene mutation (Figure 2).
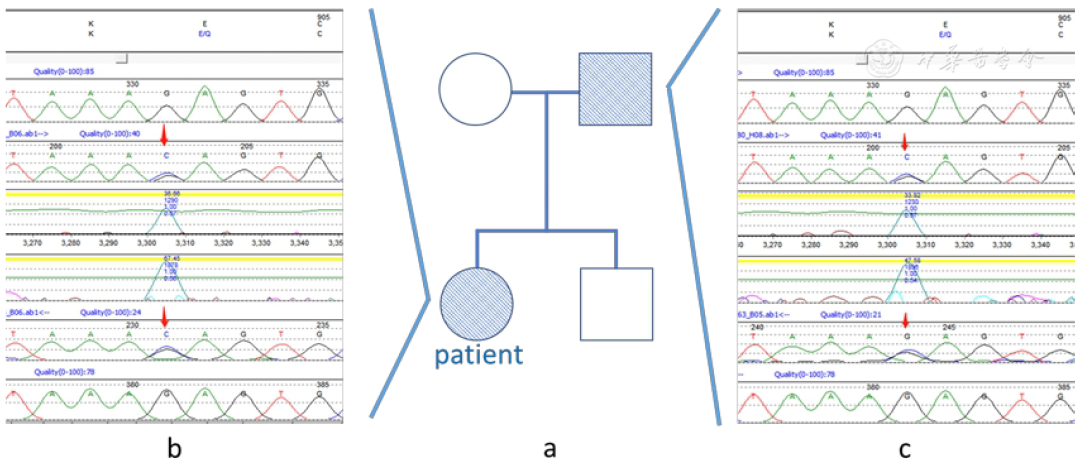
▲Figure 2 Pedigree and mutation site diagram of the patient.
Plasma fibrinogen level measured at 1.61 g/L; aspartate aminotransferase 1995.0 U/L, α-hydroxybutyric acid enzyme 665 U/L, albumin 33.9 g/L, total bilirubin 308.5 umol/L, direct bilirubin 271.7 umol/L, indirect bilirubin 36.8 umol/L. Indicating liver dysfunction; iron studies: serum iron 9.8 umol/L, ferritin 1146.3 ng/L, unsaturated iron binding capacity 38.01 umol/L, total iron binding capacity 47.8 umol/L; triglycerides 1.98 mmol/L, total cholesterol 3.34 mmol/L, high-density lipoprotein cholesterol 0.14 mmol/L, low-density lipoprotein cholesterol 1.66 mmol/L; NK cell proportion 1.7%, NKT proportion 9.5%, absolute NK cell count 20 cells/μL, total T proportion 96.0%, helper T cell proportion 10.8%, cytotoxic T cell proportion 80.2%, helper T to cytotoxic T cell ratio 0.13, double-positive T cell proportion 0.5%, double-negative T cell proportion 4.6%, total T absolute count 1155 cells/μL, absolute helper T count 130 cells/L, absolute cytotoxic T count 964 cells/μL; sputum culture: Achromobacter xylosoxidans, Klebsiella pneumoniae. Blood NGS: Human herpesvirus 5, cytomegalovirus, sequence count 2868, Enterococcus faecium, sequence count 1. Blood culture: GRE pneumonia.
CT scan of the chest and upper abdomen (Figure 3): 1. Bilateral pulmonary interstitial changes; 2. Bilateral pleural effusion with localized pulmonary atelectasis adjacent to lung tissue; 3. Multiple lymph nodes in the bilateral axillary region; 4. Enlarged cardiac silhouette; 5. Mild fatty liver with intraparenchymal calcifications; 6. Gallbladder slightly distended; 7. Splenomegaly, accessory spleen; 8. Mesenteric root haziness, bilateral renal fascia thickening, inflammatory lesions? Accompanied by multiple adjacent lymph nodes. Bedside abdominal ultrasound: 1. Low echo area in the liver—nature pending determination; 2. Gallbladder sludge-like stones; 3. Splenomegaly.

3. Diagnosis and Differential Diagnosis
① The patient is a young female with an insidious onset; ② The patient presents primarily with gastrointestinal symptoms; ③ Physical examination reveals jaundice of the skin and mucous membranes; ④ The patient has significantly elevated soluble CD25 levels, decreased NK cell activity; bone marrow examination shows significant hemophagocytosis; the patient also has a Rag1 gene mutation unrelated to hemophagocytic lymphohistiocytosis. HLH diagnostic criteria: If one or two of the following are met, the diagnosis of HLH can be confirmed (1) molecular diagnosis consistent with HLH (2) diagnostic criteria for HLH (five of the following eight criteria) (A) Initial diagnostic criteria (to be assessed in all HLH patients) fever, splenomegaly, Cytopenias (affecting ≥2 of the 3 lineages in peripheral blood): hemoglobin <90 g/L (infants <4 weeks: hemoglobin <100 g/L); platelets <100×109/L; neutrophils <1.0×109/L; hypertriglyceridemia and/or hypofibrinogenemia; fasting triglycerides ≥3.0 mmol/L (i.e., ≥265 mg/dL); fibrinogen ≤1.5 g/L, hemophagocytosis in bone marrow or spleen or lymph nodes, with no evidence of malignancy (B) New diagnostic criteria: Low or absent NK cell activity (according to local laboratory reference); ferritin ≥500µg/L; soluble CD25 (i.e., soluble IL-2 receptor) ≥2,400U/ml.
Table 1 Hscore scoring criteria: Hscore diagnostic criteria (2014), used for secondary HLH. Points are assigned for the following parameters, with higher scores indicating a greater likelihood of HLH diagnosis. The optimal cutoff value is 169, with sensitivity of 93% and specificity of 86%, allowing for accurate classification of 90% of patients.
In this case, the patient had a WBC of 0.32×109/L, RBC of 2.51×1012/L, hemoglobin of 70 g/L, hematocrit of 20.7%, and platelets of 68×109/L, indicating anemia with hemoglobin below 90g; the patient had fever with a maximum temperature of 39.5°C; splenomegaly was present; soluble CD25 level was 34445 (N<6400) significantly elevated; NK cell activity was 5.24%, indicating decreased NK cell function or number; bone marrow aspiration showed significant hemophagocytosis, meeting the HLH diagnostic criteria. Hscore score: 171 points.
4. Treatment
Primarily symptomatic treatment including anti-infection, antiviral, cough suppression, antiemetic, and liver protection. Treatment included injectable tigecycline, injectable methylprednisolone sodium succinate, piperacillin-thiabendazole, injectable ganciclovir for antiviral treatment, anti-infection, magnesium isoglycyrrhizinate injection, ademetionine for liver protection, enteric-coated rabeprazole for gastric protection, and adequate potassium supplementation with potassium chloride sustained-release tablets. Rivaroxaban tablets were administered to stabilize the cervical wall thrombus. Micafungin was given for prophylactic antifungal treatment. Oral furosemide and spironolactone were used for diuresis. Injectable doxofylline, ipratropium bromide nebulization solution, and inhaled budesonide suspension were used for cough suppression and asthma relief. Metoclopramide hydrochloride was administered for antiemetic treatment, and recombinant human erythropoietin was used to improve blood parameters.
5. Treatment Results, Follow-up, and Outcome
After treatment, the patient recovered from the disease and has been followed up to date, with all medications discontinued and complete recovery.
Discussion
Hemophagocytic lymphohistiocytosis (HLH) is a rare, fatal syndrome characterized by a strong immune activation. The most typical findings of HLH are fever, hepatosplenomegaly, and cytopenias. Other common findings include hypertriglyceridemia, coagulopathy with hypofibrinogenemia, liver dysfunction, elevated ferritin and serum transaminase levels, and neurological symptoms associated with cerebrospinal fluid abnormalities and neutrophilia.[1,2,3,4]
Reviewing the patient’s history, the patient presented with fever, vomiting, jaundice, and gastrointestinal symptoms, with liver injury as the early primary manifestation, followed by splenomegaly, trilineage cytopenias, and severe neurological symptoms such as convulsions and seizures. Bacterial culture tests did not reveal any gastrointestinal infections. Therefore, the patient had an insidious onset, with clinical manifestations appearing sequentially, typical symptoms, and significant hemophagocytosis found upon bone marrow examination, thus confirming the diagnosis of hemophagocytic syndrome. Gene testing revealed that the patient carried a heterozygous Rag1 gene mutation, with the father also having the same heterozygous mutation, but the father reported no immune abnormalities. Therefore, it is unclear whether the patient’s severe hemophagocytic syndrome is due to age, genetic abnormalities, or a combination of both.
This also serves as a reminder to young doctors and non-hematology specialists that HLH has diverse clinical manifestations, often beginning with various types of infections, with typical symptoms appearing later; if not managed promptly, it may threaten the patient’s life. HLH can often be controlled by managing symptoms; thus, when dealing with various types of severe infections, we should consider the possibility of hemophagocytic syndrome, while also providing timely symptomatic treatment to protect organ function. Furthermore, when diagnosing such diseases, especially in adolescent patients, it is essential to check the patient’s genotype and to be aware of other gene mutations that may exacerbate these symptoms, in addition to those related to hemophagocytic syndrome.
Histopathological findings include extensive accumulation of lymphocytes and mature macrophages, sometimes accompanied by phagocytosis, particularly affecting the spleen, lymph nodes, bone marrow, liver, and cerebrospinal fluid (CSF).[5] The main pathogenesis of HLH can be summarized as a disease involving a persistent immune/inflammatory response, known as a “cytokine storm”. The immune mechanism is activated by various factors such as infections, autoimmune disorders, or malignancies. After resolving these triggering factors, the immune system must be inactivated to restore normalcy. In some cases, due to abnormalities in cellular signaling pathways, inactivation cannot be achieved. Thus, a malignant activation cycle is repeated. This excessively stimulates immune cells, which then invade normal tissues, leading to organ failure and the secretion of large amounts of cytokines: interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), interleukin-10 (IL-10), and macrophage colony-stimulating factor.[6] These inflammatory cytokines contribute to the clinical features of HLH, such as bone marrow suppression, lymphadenopathy, fever, and organ dysfunction.[7] Various immunological abnormalities have been observed in HLH. The main abnormality is the defective function of cytotoxic T lymphocytes and NK cells.[8] CD8+ T cells secrete IFN-γ, which can activate macrophages.[9]
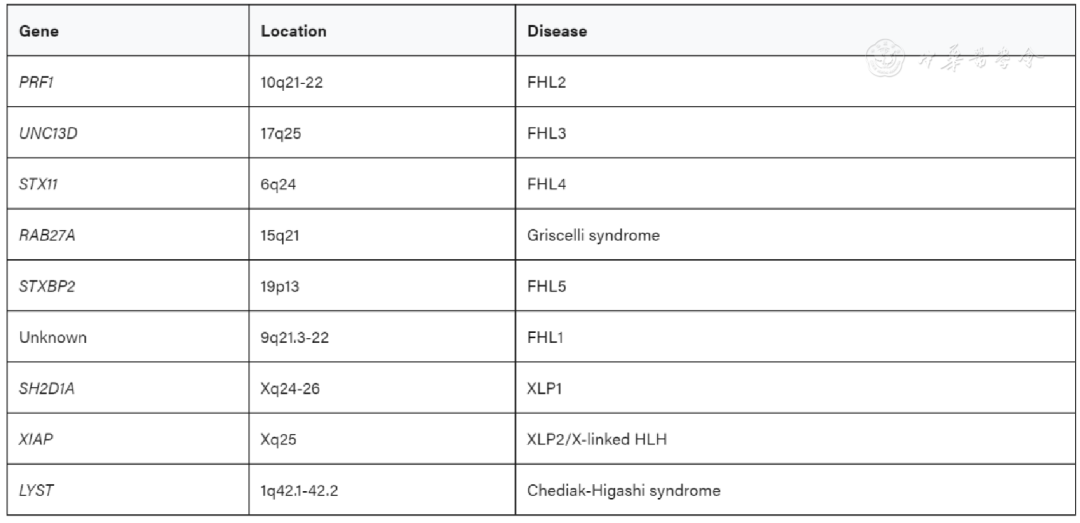
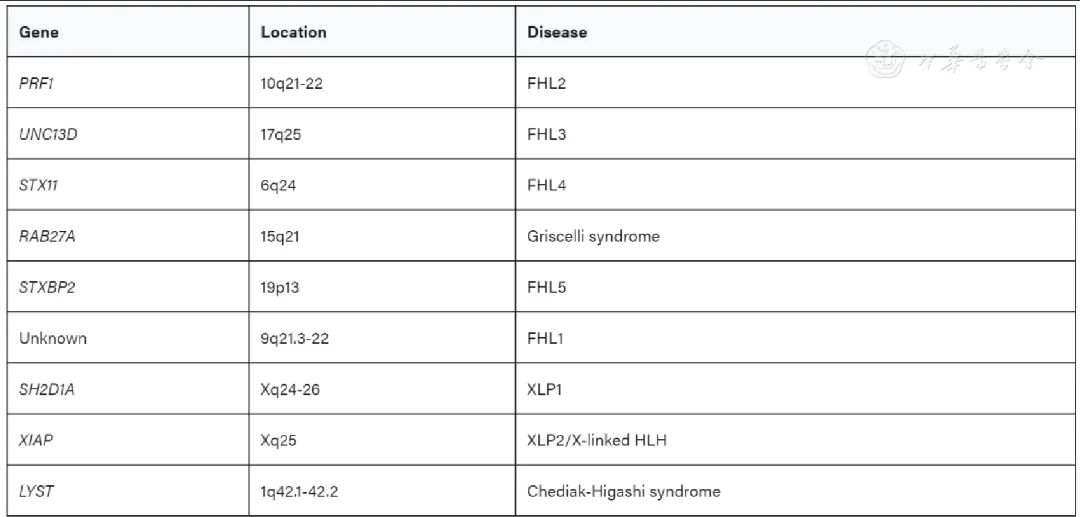
Several genes associated with HLH have been identified. Autosomal recessive mutations: PRF1, UNC13D, and STX11 encode perforin, Munc13, and Syntaxin-11, respectively, leading to FHL2, FHL3, and FHL4.[10,11,12] Mutations in the gene encoding Munc18-2, STXBP2, are the cause of familial HLH 5.[13] Hereditary mutations in RAB27A and LYST lead to immunodeficiency diseases: Griscelli syndrome type 2 (GS2) and Chédiak-Higashi syndrome type 1 (CHS1), both of which often present with HLH. Additionally, patients with Hermansky-Pudlak syndrome type 2 (HPS2) due to mutations in AP3B1 may also present with HLH. X-linked lymphoproliferative syndromes 1 and 2 (XLP1, XLP2) caused by mutations in SH2D1A or XIAP also frequently present with HLH.[14]
RAG1 expression is limited to developing T and B lymphocytes. In both lineages, there are two waves of RAG expression.[15] Loss of Rag gene expression leads to developmental stagnation at the progenitor stage of T and B cell development.[16,17] Persistent RAG expression results in abnormal thymic development and severe immunodeficiency.[18] Mutations in the RAG1 gene have been reported to be associated with Omenn syndrome, combined immunodeficiency with granulomas, or T-cell negative, B-cell negative, NK-cell positive severe combined immunodeficiency, inherited in an autosomal recessive manner. Furthermore, pathogenic variants in the RAG1 gene can lead to α/β T-cell expansion and γ/δ T-cell lymphopenia associated with severe cytomegalovirus infections and autoimmune reactions.[19]
As previously mentioned, HLH is a type of disease characterized by excessive phagocytosis due to immune system dysfunction caused by various infections, with pathogenesis primarily involving humoral immunity. The Rag1 gene mutation expressed in developing T and B lymphocytes can lead to severe immunodeficiency diseases. It is well-known that T and B lymphocytes can affect all aspects of cellular and humoral immunity; however, there have been no reports or studies indicating whether hereditary RAG1 gene mutations play a role in the onset, occurrence, and progression of HLH diseases and what specific role they may have. In this case, the HLH patient also had a hereditary RAG1 gene mutation; although this mutation did not independently cause the patient to exhibit related clinical manifestations, the role of this mutation in the pathogenesis of HLH in this patient remains to be further researched and contemplated.
(References: omitted)
Conflict of Interest StatementAll authors declare that there are no conflicts of interest in this study.
↓ Click the card below to enter the mini-program for discussion and online communication with the authors
The Case Report Mini-Program is a clinical case community for Chinese doctors.
Articles in the case library can be searched in the community and discussed with authors online.
Authors in the case library can log in to claim their articles and receive electronic inclusion certificates.
Doctor friends can publish cases they consider valuable, cases worthy of sharing in departmental meetings, lectures, or cases needing publication during teaching scenarios on the Case Report platform.
For submission of cases to the Case Report platform, please contact the editor for pre-review and assistance in submission to the library.
We welcome medical students and doctor friends to click the follow button of the Chinese Case Report public account to enter the mini-program, looking forward to your sharing and interaction~

Previous Issues · Recommended
Case Report: Microwave Ablation Treatment of Pulmonary Aspergillosis
Case Report: Microwave Ablation Treatment of Pulmonary Aspergillosis
Case Report: PKLR Gene Mutation Causing Severe Pyruvate Kinase Deficiency
Case Report: Schnitzler Syndrome Presenting as Clubbing Finger