
Lu Xingguo Ye Xiangjun
(2)T−B− SCID
1. Recombinase-activating Genes1 and 2 Deficiency Recombinase-activating genes1 and 2 (recombinase-activating genes 1 and 2,RAG1/2) deficiency accounts for approximately6% of SCID patients. In about75% of patients with RAG1/2 deficiency, there are very low numbers of T, B lymphocytes, and NK cells. This type of SCID is caused by mutations in the products encoded by the RAG1 and RAG2 genes—mutations occur in one of the two DNA binding proteins. These proteins play a critical role in guiding the somatic rearrangement of immunoglobulin and T cell (antigen) receptors (T cell receptor,TCR). RAG1 and RAG2 proteins bind to the side wings of specific recognition sequence gene segments, participating in the cutting and splicing assembly of mature immunoglobulin or T cell receptor (TCR) gene DNA (Figure 9-2). B and T cell precursors are not lacking but are stalled at an early stage of development. Some patients with mutations in RAG1 or RAG2 have varying degrees of immunodeficiency and atypical lymphocyte phenotypes, with a few having limited development of B or (and) T lymphocytes. In the absence of other specific molecular diagnoses, mutations in these genes should be sought in some immunodeficient individuals.
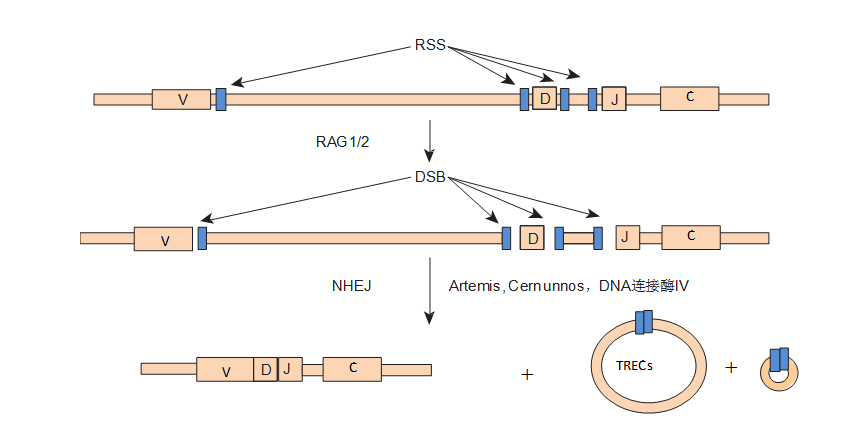
Figure9-2 Gene Defects Affecting Lymphocyte Antigen Receptor Gene Rearrangement, Resulting inT−B−NK+ SCID.
The assembly of immunoglobulin and TCR is formed from subunit segments separated in the germline and undergoes reassembly or somatic rearrangement during lymphocyte development. The figure shows the TCR or immunoglobulin heavy chain gene. The heavy chain consists of four different parts: variable region (variable, V), diversity region (diversity, D), joining region (joining, J), and constant region (constant region, C). Some of these segments flank the recombination signal sequences (recombination signal sequence, RSS). RAG1 and RAG2 initiate the process that leads to double-strand breaks (double-strand breaks, DSB) in the DNA. The DNA between the coding segments is looped out, and the coding sequences are joined into new segments. In T cells, the larger DNA loops are referred to as T cell receptor excision circles (T cell receptor excision circles, TREC). All these DNA connections are executed by a set of common molecules that promote nonhomologous end-joining (nonhomologous end-joining, NHEJ) function.
Newborn screening for SCID can be performed by extracting DNA from dried blood spots and detecting TREC using methods such as real-time quantitative PCR (RT-PCR). 70% of T cells generate δRec-φJαTREC in late maturation. TREC is very stable in T cells, with the highest levels in newborns (TREC>105 copies/μL), while SCID patients show extremely low levels of TREC.
2. DCLRE1C (Artemis) Deficiency is a recombinase system that participates in every step of the V(D)J rearrangement reaction and is composed of multiple proteins. Nine known proteins are involved in this process. The DCLRE1C gene encodes the Artemis protein, which plays a role in opening the hairpin structure in the second step of the reaction (Figure 9-2).
3. DNA-PKcs Deficiency DNA -dependent protein kinase (DNA-dependent protein kinase, DNA-PKcs) is a nuclear serine/threonine protein kinase belonging to the phosphatidylinositol 3-kinase-related kinase family (phosphatidylinositol 3-kinase related kinase, PIKK), and is the most important protein factor involved in NHEJ repair. It has three subunits: one catalytic subunit and two DNA binding subunits—Ku70/Ku80 heterodimer. When DSB occurs, the two broken DNA ends each bind a Ku70/Ku80 heterodimer, and the two dimers further bind to form a “bridge” structure (Ku ring) at the break, recruiting DNA-PKcs molecules to bind with DNA. Additionally, the inner side of the Ku ring is lined with positively charged amino acids, which can interact with the negatively charged DNA strand, fixing the DNA double helix at a specific position, facilitating the proximity and connection of the DNA break ends.
4. Lymphoid Hypoplasia and Mitochondrial Adenylate Kinase2 Deficiency Lymphoid hypoplasia is the most severe SCID caused by mutations in the adenylate kinase 2 (adenylate kinase-2, AK2) gene. It is primarily characterized by severe reductions in peripheral blood neutrophils and lymphocytes, accompanied by thymic hypoplasia, often leading to bilateral sensorineural hearing loss in newborns, with lethal infections occurring within days to months after birth.AK2 gene is mainly expressed in the inner mitochondrial membrane and regulates the levels of adenosine diphosphate. Deficiency of AK2 leads to increased apoptosis of myeloid and pre-lymphoid cells.
5. Adenosine Deaminase and Purine Nucleoside Phosphorylase Deficiency Adenosine deaminase (adenosine deaminase, ADA) and purine nucleoside phosphorylase (purine nucleoside phosphorylase, PNP) deficiencies are classified as related to T−B−NK−SCID in the 2011 IUIS Primary Immunodeficiency Disease Classification by the Expert Committee. In the 2014 version of the classification, the latter condition was classified as “less severe than severe combined immunodeficiency,” but due to the strong correlation between the two, they are discussed together here. Both enzymes are involved in the salvage pathways of purine nucleotide synthesis (Figure 9-3), resulting from mutations in the genes ADA and PNP. Deficiencies in ADA and PNP are the only known immunodeficiencies related to nucleotide synthesis. ADA is a purine degrading enzyme that removes an amino group from adenosine to produce inosine; the gene is located on chromosome 20q13, with a cDNA length of 1533 bp, encoding 362 amino acids; most gene mutations are missense mutations. The incidence of ADA deficiency (ADA-deficient, ADAD) is 1 in 20,000 live births, accounting for about 15% of SCID patients, and about one-third of autosomal recessive SCID cases.
PNP deficiency (PNP-deficient, PNPD) is very rare. ADA or PNP deficiencies lead to the accumulation of intracellular deoxyadenosine and deoxyguanosine. These molecules are not toxic to lymphocytes themselves. However, when converted to 5′ triphosphates by deoxyadenosine triphosphate (deoxyadenosine triphosphate, dATP) and deoxyguanosine triphosphate (deoxyguanosine triphosphate, dGTP), they can inhibit ribonucleotide reductase, preventing the de novo synthesis of deoxynucleotides. Without the materials to replicate and repair DNA, cells stop dividing, and lymphocytes are more susceptible to death than other cell types. T precursor cells in ADAD and PNPD appear to be particularly sensitive to apoptosis (programmed cell death). In ADAD, B cell depletion is more common than in PNPD. In the latter, the B cell phenotype is more variable.
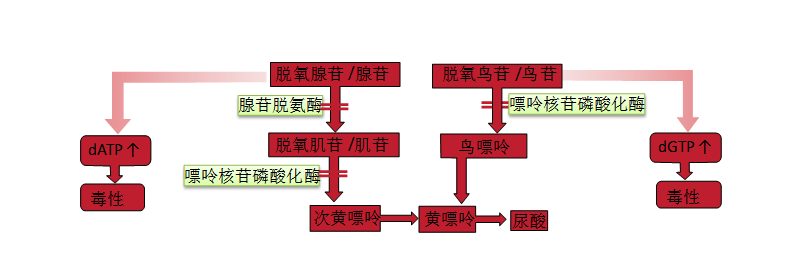
Figure9-3Salvage Pathway of Purine Nucleotide Biosynthesis
About 85% of ADAD patients exhibit significant reductions in T and B cell numbers and low serum antibody levels, presenting a SCID phenotype. In ADAD patients, NK cells are rarely seen. Other clinical manifestations may include imaging changes in the ribs, vertebrae, and iliac crests, as well as subsequent cognitive impairments. Without bone marrow transplantation, most patients die from infections. 10% to 15% of ADAD patients may present with delayed or late-onset forms, which may manifest in late infancy or early childhood. These patients may initially have varying numbers of circulating lymphocytes and some humoral immunity, which rapidly diminish. The severity of the phenotype seems to be somewhat associated with residual ADA activity. Residual enzyme activity of 3% to 5% may be sufficient to maintain normal immune function, and even extend into adulthood, where infectious complications may occur. Residual ADA activity has also been found in several adult “idiopathic” CD4 lymphopenia patients.
PNPD primarily presents as selective cellular immunodeficiency. Due to its biochemical pathophysiology being similar to that of ADAD, it is often discussed in the literature together with ADAD. Patients have reduced circulating T cell numbers and are highly susceptible to viral and fungal infections. Initially, patients often have normal B cell numbers and normal serum immunoglobulin levels. Over time, humoral immunity tends to deteriorate. Analyses of mouse models of PNPD indicate that dGTP accumulation inhibits mitochondrial DNA repair and leads to increased T cell apoptosis. Approximately 50% of PNPD children exhibit neurological complications such as spasticity, bilateral paralysis, progressive dementia, and other motor disorders, including cognitive impairments.
For clinically suspected cases, diagnosing ADAD or PNP is relatively straightforward. The activity of ADA and PNP can be easily measured in red blood cell or white blood cell lysates. In symptomatic patients, the activity is usually only 1% or less than that of normal subjects. These methods can also be used for screening in amniotic and chorionic villi cultures.PNP is required for the generation of uric acid, which is a precursor for hypoxanthine (Figure 9-3). Therefore, individuals with PNPD have decreased serum and urinary urate levels, while ADAD patients do not.
ADAD is effective for enzyme replacement therapy. Red blood cells contain high levels of ADA, and red blood cell transfusion can improve the enzyme deficiency. About 50% of patients respond to this treatment, mainly those who still have residual immune function. These patients are at risk for long-term complications from red blood cell transfusions, such as iron overload and transfusion-related infections. Another treatment option is the infusion of polyethylene glycol-conjugated bovine ADA (polyethylene glycol–conjugated bovine ADA, PEG-ADA or methoxypolyethylene glycol succinimidyl adenylate deaminase). This therapy is generally well tolerated, but immune reconstitution is incomplete. During PEG-ADA treatment, infectious complications and malignancies such as EB virus-positive lymphoma may occasionally occur. Red blood cell transfusions are of no benefit for PNPD patients. Other forms of enzyme replacement therapy have not yet been developed.
Bone marrow transplantation can cure ADAD and PNPD related immunodeficiencies. After transplantation, characteristics such as neurological deficits cannot be improved. ADAD can also be cured by gene therapy.
(3)Others
Include some combined immunodeficiency diseases that are usually less severe than severe combined immunodeficiency, as detailed in Table 9-1.
SCID infants with maternal T cell transplantation may have dysfunctional T cells, leading to autoimmune cytopenias or graft-versus-host disease. In some genes causing SCID, suballelic mutations can lead to Omenn syndrome (OS) or “leaky” SCID. In this case, patients may have higher T cell numbers, but their functional activity is not entirely absent as in typical SCID, but rather reduced. Different clinical manifestations can be seen in typical SCID, OS, leaky SCID, granulomatous disease with T cell reduction, etc., all of which can be attributed to mutations in the RAG gene. Defects in the RAC2 gene lead to leukocyte motility defects, as shown in Table 9-5; in one case of RAC2 deficiency, TCR excision circle (TREC) defects were found during newborn screening, but T cell numbers and responses to mitogen were normal. Other diseases associated with T cell reduction, such as DNA repair disorders, cartilage-hair hypoplasia, IKAROS defects, and NEMO syndrome, are shown in Tables 9-2 and 9-6, and the severe forms of these diseases should exhibit clinical symptoms and signs of SCID. Severe folate deficiency (e.g., due to absorption disorders caused by folate transporter and transport genes SLC10A1 or PCFT defects) and some metabolic diseases, such as methylmalonic aciduria, can also present with reversible lymphocyte reduction, in addition to their characteristic manifestations.
II. Combined Immunodeficiency Diseases with Syndromic Features
There are 16 types of combined immunodeficiency diseases with syndromic features, as detailed in Table 9-2.
Table9-2 Combined Immunodeficiency Diseases with Syndromic Features
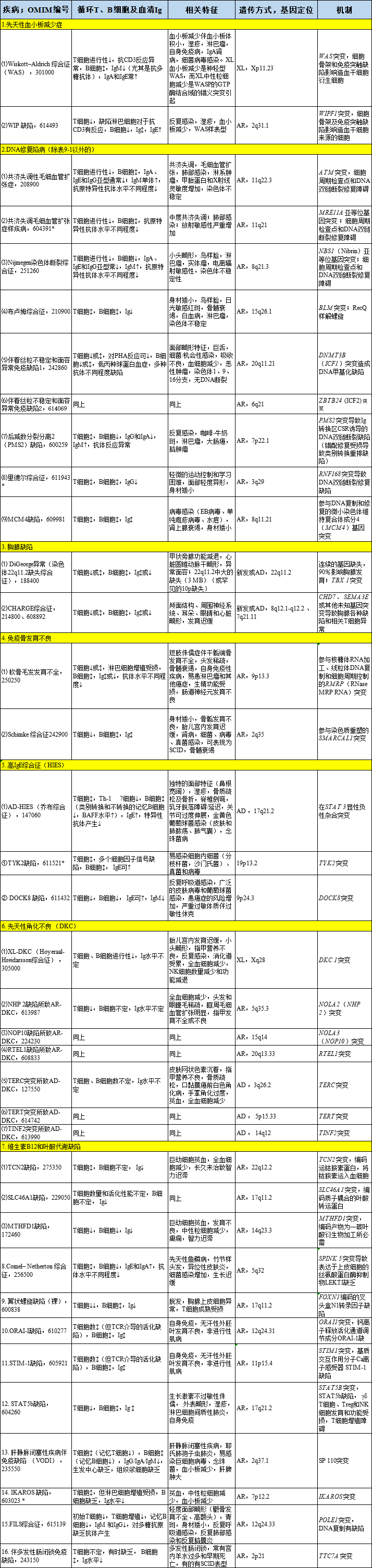
XL denotes X-linked inheritance; AR denotes autosomal recessive inheritance; AD denotes autosomal dominant inheritance; SCID denotes severe combined immunodeficiency disease; DOCK8 denotes cytoplasmic division factor protein 8. Progressive ↓ represents progressive decline, ↓ represents decline, ↑ represents normal or increased, ↕ represents normal, ↑ represents increased.* Fewer than 10 cases have been reported in the literature to date. The T and B cell numbers and functions of these diseases exhibit a wide range of abnormalities; the most severe cases meet the diagnostic criteria for SCID or leaky SCID and require immune system reconstruction treatment, such as allogeneic hematopoietic stem cell transplantation. Not all patients with DOCK8 deficiency have elevated serum IgE, but most have recurrent viral infections and malignancies due to combined immunodeficiency. AR-HIES caused by TYK2 deficiency is also seen in association with atypical mycobacterial disease leading to MSMD, as detailed in the following Table 9-6. Riddle syndrome is caused by mutations in genes involved in DNA double-strand break repair and is associated with hypoglobulinemia. Autosomal dominant and recessive forms of congenital ichthyosis are included in this table. Lastly, an isolated case of a premature infant who died on day 87 was found to have an IKAROS defect. Patients exhibit a lack of B cells and NK cells and non-functional T cells.
