
“
Click the blue text to follow us

Li Guobo

Professor at West China School of Pharmacy, Sichuan University, PhD supervisor, and research group leader. Mainly engaged in drug design and medicinal chemistry, focusing on the design and discovery of drugs targeting metal enzymes. He has led several research projects, including the National Excellent Youth Fund Project, the National Natural Science Foundation’s general project, and international cooperation projects in Sichuan Province. As the first or corresponding author, he has published over 40 SCI papers in journals such as Chem Sci, J Med Chem, Bioinformatics, Med Res Rev, Acta Pharm Sin B, Drug Discov Today, and J Chem Inf Model. He has applied for more than 10 national invention patents, with 3 granted patents and 5 granted software copyrights. He has received several awards, including the First Prize in Natural Science from the Ministry of Education, the First Prize in Natural Science from the Sichuan Provincial Science and Technology Progress Award, the Sichuan University Academic Newcomer Award, and the Excellent Scholar Award from Sichuan University. He serves as a guest editor for Eur J Med Chem and as a young editorial board member for the bilingual special issue of Chinese Journal of Pharmacy and Chinese Chemical Letters.
Exclusive Original | Li Guobo: Understanding Deep Learning Technology in Drug Discovery PPS
Dai Qingqing, Yu Junlin, Li Guobo*
(Department of Medicinal Chemistry, West China School of Pharmacy, Sichuan University, Chengdu 610041, Sichuan, China)
[Abstract] Deep learning technology has achieved significant breakthroughs in recent years and has been applied in various fields such as medicine and pharmacy. This article focuses on the development and application of deep learning in innovative drug discovery, providing a detailed review of representative cases where deep learning has been used for protein structure prediction, drug target prediction, drug-target interaction prediction, drug synthesis route design, de novo drug molecule design, as well as ADMET (absorption, distribution, metabolism, excretion, and toxicity) prediction. It also summarizes the challenges faced by existing methods and potential solutions, aiming to provide insights and considerations for the development and application of deep learning-assisted methods in drug discovery.
The concept of artificial intelligence (AI) originated in 1956 and, after half a century of exploration, entered a period of rapid development in 2011, becoming a new technological science that is pushing humanity into the intelligent era. Deep learning (DL), also known as deep neural networks, is a popular research direction in the field of AI. It processes and abstracts sample data through multiple layers of nonlinear information to uncover intrinsic patterns, addressing problems such as feature learning, classification, and pattern recognition. Current mainstream DL models include convolutional neural networks (CNN), recurrent neural networks (RNN), and graph neural networks (GNN), along with their variants such as deep residual networks (ResNet), variational autoencoders (VAE), adversarial autoencoders (AAE), generative adversarial networks (GAN), and message passing neural networks (MPNN). These DL models have achieved unprecedented success in fields like image recognition, speech recognition, machine translation, human-computer games, and autonomous driving, profoundly changing people’s production and lifestyle[1-2].
At the same time, DL technology has gradually emerged in fields such as medicine, pharmacy, and life sciences. For example, in 2018, the Waller team extracted chemical transformation rules from 12.4 million single-step reactions using a DL network and developed a new algorithm that combines three different neural networks with Monte Carlo tree search, achieving efficient design of compound synthesis routes[3]. Subsequently, the Jensen and Jamison teams reported a platform that integrates synthesis route design and automated synthesis, completing the automated synthesis of 15 small molecule drugs, further advancing the field[4]. Recently, the Hassabis team reported a new protein structure prediction tool, AlphaFold2, which has greatly improved the accuracy of protein structure prediction by integrating physical and biological knowledge into DL methods[5]. Meanwhile, the Baker team also reported a new protein structure prediction tool, RoseTTAFold[6], which employs an attention mechanism to allow the entire DL to learn information across different dimensions of protein primary, secondary, and tertiary structures, achieving prediction accuracy comparable to AlphaFold2. In recent years, several DL methods have also been developed for predicting drug-target interactions, drug target predictions, de novo drug design, and predicting drug properties (mainly including ADMET), serving multiple important aspects of innovative drug development. These tools may change the process of innovative drug development and improve drug development efficiency. Therefore, this article focuses on the development and application of DL in innovative drug discovery, reviewing representative DL cases and research ideas, summarizing their application characteristics, challenges faced, and possible solutions, hoping to provide references and considerations for the development of DL in the field of drug discovery.
1
Protein Structure Prediction Based on Deep Learning
The three-dimensional structure of proteins is an important foundation for studying drug target functions and drug design. How to quickly and efficiently obtain accurate protein structures is a scientific problem that needs to be solved. In the early stages, researchers predicted protein three-dimensional structures based on statistical protein evolutionary information, using traditional machine learning methods (such as Monte Carlo methods, support vector machines, etc.) and fully connected neural network (FNN) models. For example, Bohr et al.[7] and Fariselli et al.[8] used target protein primary sequences, homologous protein sequences, and associated mutations to train FNN models to predict the backbone structure of proteins, but there was still a significant gap in achieving precise predictions of protein three-dimensional structures.
As protein structure data continues to increase and DL technology rapidly develops, more complex deep network models and richer protein sequence information are applied to predict protein three-dimensional structures, breaking through the bottleneck of directly obtaining protein three-dimensional structures from primary sequences, with prediction accuracy approaching experimental resolution levels. The protein structure prediction based on DL has been a direction that researchers have been attempting and striving for. The general process involves obtaining evolutionarily related multiple sequence alignment (MSA) features through sequence alignment, combining protein sequence encoding as input, using deep network models to predict contact maps between residues or more specific distance distributions, as well as the dihedral angle distributions of protein backbones, and then reconstructing the protein three-dimensional structure with the predicted spatial structural information as constraints (see Figure 1). For example, the latest protein structure prediction tool AlphaFold2 reported by the Hassabis team achieved the best prediction ranking in the recent protein structure prediction technology assessment (i.e., The 14th Edition of Critical Assessment of Structure Prediction, CASP14), with a median score of 92.4 in the global distance test (GDT), reaching experimental resolution levels. AlphaFold2 is a neural network model based on attention mechanisms, consisting of Evoformer network modules and structure generation modules, which directly generates the three-dimensional structure of proteins from given primary sequences while learning the physical and biological knowledge of protein structures. Baek et al.[6] also developed a new end-to-end protein structure prediction tool, RoseTTAFold, based on attention mechanisms. This tool is a three-track network model, using progressively connected networks to transfer and process information from protein primary, secondary, and tertiary structures, with multiple connections between tracks allowing the network to simultaneously learn the relationships between sequences, inter-residue distances, and atomic coordinates. Experimental results show that RoseTTAFold not only achieves prediction accuracy close to AlphaFold2, providing explanations for the biological functions and mechanisms of unknown structure proteins, but also quickly constructs accurate protein-protein complex structures based on sequence information. In terms of required computational resources and time, RoseTTAFold also shows certain advantages over AlphaFold2; excluding the time spent on sequence alignment and template search, it can generate the protein 3D backbone structure within 10 minutes using only one graphics processing unit (GPU). Additionally, Rahman et al.[9] improved upon the ResNet model to propose a DL model for predicting inter-residue distances, achieving high-precision predictions of true distances between protein residues using fewer protein features, including two co-evolutionary features and three non-evolutionary features, with an average score improvement of over 10% compared to state-of-the-art methods, providing a new reference for protein structure prediction.
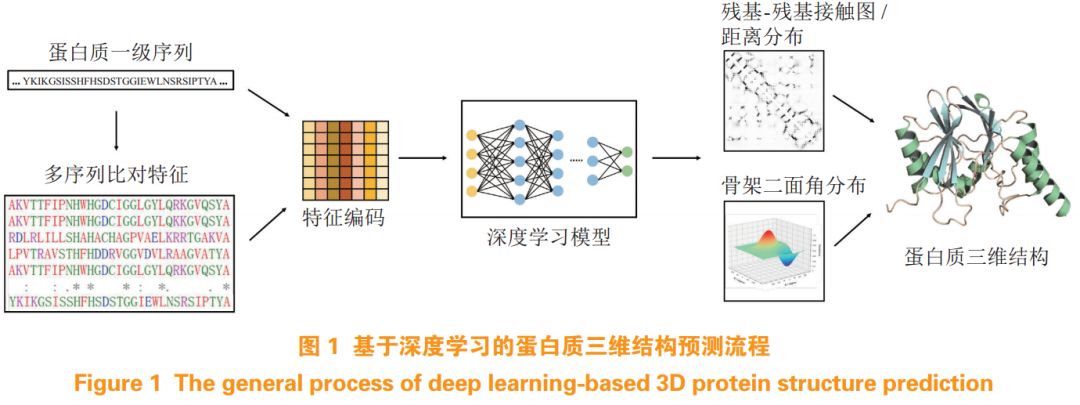
Furthermore, Yang et al.[10] first proposed using a GAN model to predict protein residue-residue contact maps, demonstrating good predictive performance on benchmark datasets. This model is named GANcon; it trains the generative model and discriminative model through adversarial learning strategies, ultimately generating contact maps close to the real data distribution. The generative model employs an encoder-decoder framework to capture potential inter-residue contact information from various protein sequence features, generating realistic residue contact maps; the discriminative model uses a CNN based on residue blocks as input to the generated or real contact maps—protein sequence feature samples—to identify differences between the generated contact maps and real contact maps, driving the generative model to produce more accurate contact maps. They also introduced a new symmetric focal loss function to address the data imbalance issue within contact maps. However, GANcon still has room for improvement in terms of training stability and input feature selection.
2
Drug-Target Interaction Prediction Based on Deep Learning
Drug-target interaction (DTI) is a crucial foundation for drug discovery. Accurate and effective DTI predictions can greatly assist drug R&D and accelerate the discovery of lead or candidate compounds. In recent years, methods for predicting DTI based on DL have been reported, with a general workflow illustrated in Figure 2, where researchers construct distinctive descriptors based on the structure and physicochemical properties of drugs and targets and employ different DL network models to learn DTI patterns, ultimately predicting the likelihood or interaction strength of DTI.
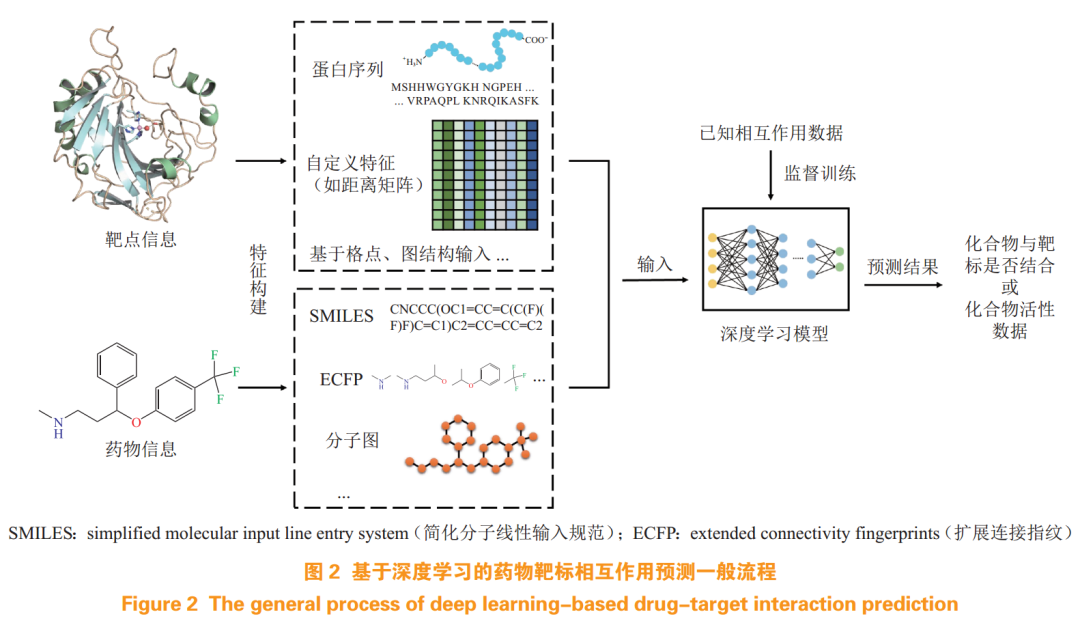
Early researchers tended to use simple, direct input data and a single-structure network framework. For instance, drug structure information and target sequence information were employed to learn interaction features through basic versions of RNN, CNN, etc.[11-12] However, the predictive results were not ideal. Researchers found that merely applying drug-target-related information using DL models could not fundamentally resolve the issue; it was necessary to reasonably construct input descriptors based on the dual theoretical guidance of DL and drug discovery, considering various properties of drugs and targets, and to build neural network frameworks adapted to the drug-target system to effectively enhance the model’s predictive ability and result reliability. On this basis, a series of DL networks based on grids, graph structures, and new algorithms were developed, reasonably incorporating attention mechanisms and other algorithms to enhance model interpretability.
The grid-based feature construction method contains richer spatial information, making it more suitable for DTI prediction systems. Features constructed using this method can be viewed as a three-dimensional image, which can be trained and learned using three-dimensional CNN models, though it faces issues such as large parameter counts and high computational costs. Li et al.[13] borrowed lightweight three-dimensional CNN models like ShuffleNet and Xception to construct the DeepAtom model for predicting drug-target affinity. Besides possessing various advantages of three-dimensional CNN models, the DeepAtom model also addresses the excessive parameters issue of three-dimensional CNN models through depthwise separable convolutions, using multiple small convolution kernels instead of a single large convolution kernel to reduce parameters while increasing network complexity. The model achieved a Pearson correlation coefficient of 0.831 on the core test set of PDBbind (2016 version), demonstrating strong predictive capability.
Zheng et al.[15] have a different understanding of DTI prediction; they abstract DTI prediction as a visual question answering (VQA) problem, utilizing drug SMILES and target residue distance matrices as input, and constructed the DrugVQA model based on CNN and RNN models, introducing attention mechanisms to enhance model interpretability. After training and hyperparameter optimization, the DrugVQA model exhibited exceptional predictive capability on the DUD-E database, achieving an area under the receiver operating characteristic curve (ROC-AUC) of 0.972.
GNN models have also gained attention in this field. Cho et al.[16] proposed a special GNN model, introducing the InteractionNet framework to predict binding constants between drugs and targets. The InteractionNet model is an unconventional GNN model that considers both covalent and non-covalent interactions when modeling the drug-target system. Finally, based on the PDBbind dataset, it was validated using a 20-fold cross-validation method, achieving a root mean square error (RMSE) of 1.321, outperforming the PotentialNet model (RMSE of 1.343).
Zeng et al.[17] believe that simply concatenating the feature vectors of drugs and targets to represent their interactions cannot accurately describe the complex interaction system between them, necessitating a special algorithm or network to resolve this. Accordingly, they proposed a multi-attention module MATT_DTI, which first extracts the inter-atomic relationships of the drug through a relative self-attention module, learns the hidden information of the drug and target separately using CNN modules, and finally extracts interaction information and provides predictive results through multi-head attention modules and fully connected layers. This method performed well on the KIBA and Davis datasets, consistently outperforming similar models in predictive effectiveness; for instance, when tested using the KIBA dataset, the MATT_DTI model achieved a mean squared error (MSE) of around 0.15, lower than other benchmark models’ MSE metrics. Sajadi et al.[18] constructed an unsupervised denoising autoencoder (DAE) model using drug fingerprint matrices and drug-target matrices as input, naming it AutoDTI++. This method achieved an ROC-AUC value of 0.85 when predicting random drug-target pairs on the G protein-coupled receptor (GPCR) dataset, showing a significant improvement compared to similar algorithm models.
3
Drug Target Prediction Based on Deep Learning
Drug target prediction can help researchers identify potential targets for known drugs or active molecules, thereby facilitating drug repurposing, toxicity prediction, and other applications. The aforementioned DTI prediction methods can also be utilized for drug target prediction. In addition, DL methods based on heterogeneous networks have also been applied to drug target prediction, characterized by utilizing multi-dimensional information (see Figure 3) such as drug-disease information, target-target information, and drug-target information as input features for the network, which are further transformed into a feature matrix that can be processed by a set of DL models to achieve drug target predictions.
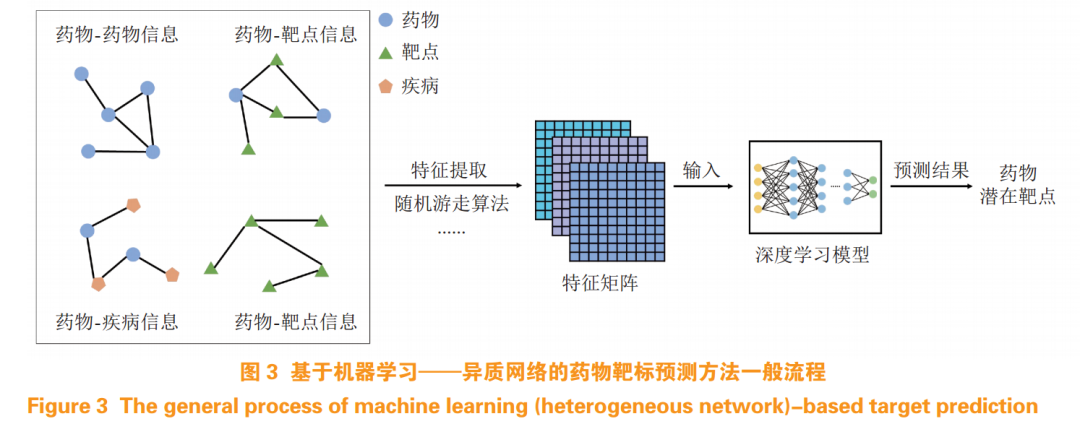
Autoencoders (AE) and their variants, such as DAE, are mainstream in heterogeneous network-based target prediction methods. Researchers collect various drug and target-related information to construct heterogeneous networks, using various AE variants for learning, ultimately analyzing and predicting potential drug targets. Zeng et al.[19] collected drug-disease, drug-adverse reaction, drug-target, and drug-drug related information to construct heterogeneous networks, extracting relationships between drugs and targets, calculating probabilistic co-occurrence matrices (PCO) using random walk algorithms, and then calculating positive pointwise mutual information (PPMI) matrices to represent the overall structure of the heterogeneous network, which is used to train DL network models, developing the deepDR model. Compared to benchmark models, deepDR achieved better predictive performance, with an ROC-AUC of 0.908. Later, they further improved the model[20], designing a new model (deepDTnet) that optimized input and framework aspects, enriching the information contained in heterogeneous networks, adding more target-related information such as target-target similarity and target-disease information, while retaining the representation of PCO and PPMI matrices, employing multi-layer DAE to learn the hidden information of heterogeneous networks. Compared to deepDR, deepDTnet demonstrated stronger predictive capabilities, achieving an ROC-AUC of 0.963. Other researchers have attempted to develop new network models by combining AE with other network models. For example, Peng et al.[21] proposed the DTI-CNN model, which is characterized by using the Jaccard similarity coefficient combined with the random walk with restart (RWR) algorithm to extract drug and target features, and added a CNN module after the DAE layer to predict final results, achieving an ROC-AUC of 0.9416 after training, comparable to deepDTnet’s performance.
Besides AE and its variants, other models have also shown promising predictive results in drug target prediction. Manoochehri et al.[22] utilized simpler inputs (considering only drug-drug similarity and target-target similarity information) and FNN models for learning predictions, but focused more on input data processing, proposing unique feature extraction and construction methods. They utilized the topological structure of heterogeneous networks to predict unknown targets of drugs, abstracting the drug-target heterogeneous network into a semi-bipartite graph and extracting multiple closed subgraphs, then applying the Weisfeiler-Lehman algorithm to rank and label nodes within each subgraph to represent the topological structure of drug-target pairs. Finally, they trained the FNN model using this special input, performing 10-fold cross-validation. The results showed that this method outperformed similar models such as BLMNII, CMF, and HNM in predictive ability. Additionally, GNN models have been used to process these heterogeneous networks for drug target prediction. Huang et al.[23] proposed the SkipGNN model, positing that directly connected nodes in heterogeneous networks may not have strong similarities, while indirect or skipped nodes may be more relevant. Based on this idea, they constructed a heterogeneous network using drug-drug, target-target, drug-target, and gene-disease related information to extract skipped similarity information and build a skip interaction graph, integrating it with the original graph input to the GNN model, ultimately outputting the probability of drug-target interactions via a decoder. Experimental results indicated that the SkipGNN model outperformed other models, such as DeepWalk, graph convolutional networks (GCN), and node2vec models.
4
Synthesis Route Design Based on Deep Learning
Drug development relies heavily on synthesis route design; designing efficient synthesis routes can significantly reduce drug development costs, shorten production cycles, and improve drug development efficiency. Traditional computer-aided synthesis route design methods mainly rely on a large number of “expert” rules and retrosynthetic analysis methods to plan synthesis routes. However, these methods often suffer from slow design speeds and often unreasonable synthesis routes[24]. With the demonstrated potential of DL algorithms in predicting compound properties and biological activities, they have gradually been applied to synthesis route design and have made certain progress.
The Waller team reported an AI tool, 3N-MCTS, in 2018 that uses three different deep neural networks (namely, expansion policy networks, selection networks, and demonstration policy networks) combined with Monte Carlo tree search algorithms to design synthesis routes for target compounds[3]. They first utilized the expansion policy network to perform reverse chemical transformations on target molecules, searching for possible transformation paths at the current node, then used the selection network to analyze and determine whether reactions were feasible, filtering out unreasonable reaction routes, and finally employed the demonstration policy network for multiple random sampling to evaluate and score the search nodes. Researchers trained these networks using 12.4 million reaction data from the Reaxys database to learn chemical transformation rules. Compared to other methods, 3N-MCTS significantly improved synthesis route search speed and quality, capable of generating hundreds of synthesis routes for compounds in a short time, with double-blind experimental results indicating that 3N-MCTS’s predicted molecular synthesis routes are close to the level of synthetic chemists. The advantage of this method lies in its ability to learn known reaction transformation rules without requiring expert-defined rules, allowing DL models to quickly select the best synthesis routes based on learned rules.
Subsequently, Coley et al.[4] launched an AI-based automated synthesis platform, initially generating synthesis routes for target molecules using feedforward neural networks, then executing a series of specific preparation processes based on the synthesis plans through robots, achieving automated synthesis. The researchers trained the network model using reaction data from the Reaxys and USPTO databases to learn reaction transformation rules, designing feasible synthesis routes for target compounds, including specifying reaction conditions, while further filtering to obtain optimal synthesis routes based on whether the reaction types are easily realizable and the diversity of intermediate products. Ultimately, they successfully completed the synthesis route design for 15 small molecule drugs and achieved automated synthesis through this platform. Concurrently, the development of DL-based sequence-to-sequence (seq2seq) models (such as transformer models) has provided a new solution for template-independent retrosynthetic prediction tasks (see Figure 4): this task can be viewed as a machine translation task in natural language processing (NLP), inputting the SMILES sequence of the target molecule and outputting the corresponding single-step reactant SMILES sequence without relying on reaction rules.
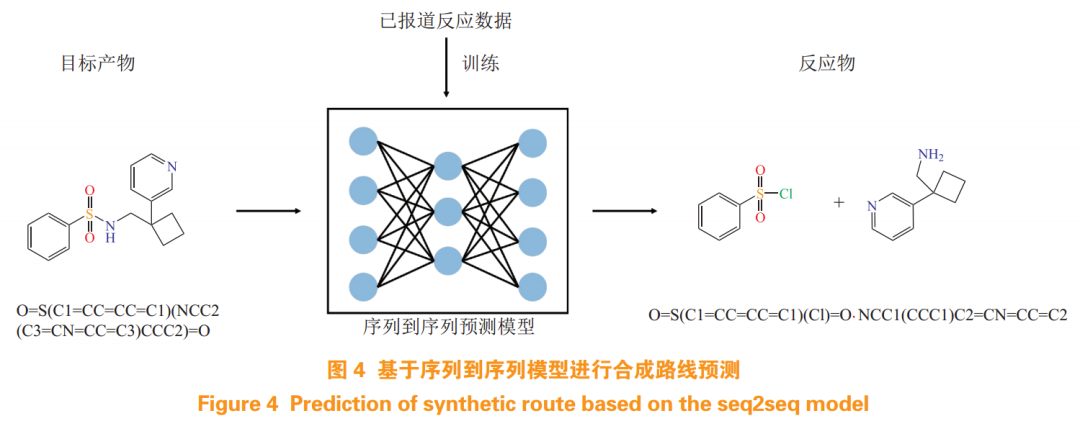
Liu et al.[25] were the first to apply the seq2seq model to retrosynthetic prediction tasks, using an RNN-based encoder-decoder structure trained on a dataset containing 50,000 patented reactions, achieving initial results comparable to rule-based benchmark methods. This method breaks through the limitations of expert rules to some extent and shows good scalability advantages. Subsequently, the seq2seq model evolved into the more popular transformer model based on attention mechanisms. Zheng et al.[26] developed a template-free self-correcting retrosynthetic route prediction tool, SCROP, predicting retrosynthetic routes using a transformer network model based on multi-head attention mechanisms, while integrating a transformer-based syntax corrector to amend unreasonable candidate precursor molecule SMILES generated by the prediction model. SCROP achieved a prediction accuracy of 59% on benchmark datasets, improving by 6% compared to template-based methods; experimental results indicated that the addition of the syntax corrector enhanced the prediction quality of the model, reducing the proportion of invalid candidate precursor molecules from 12.1% to 0.7%. Additionally, Guo et al.[27] combined the transformer model with Bayesian inference algorithms for retrosynthetic prediction, treating the task as a combinatorial optimization problem to find the optimal set of reactant pairs from all available reactant combinations to synthesize the target product. They first used the trained MolecularTransformer model to make high-precision forward predictions on given reactant combinations, then inverted the forward prediction model into a retrosynthetic model based on Bayesian theorem, exploring the best reactant combinations using Monte Carlo search algorithms. The combination of forward and backward prediction models improved the feasibility of synthesis routes while addressing the ill-posed nature of retrosynthetic problems.
This type of sequence model generally uses the SMILES strings of molecules as input, failing to effectively characterize the complex relationships between atoms in molecules. To address this, Shi et al.[28] proposed a template-free retrosynthetic prediction framework based on graph neural networks, G2G (graph to graph framework), utilizing graphs to represent molecules and transforming the task into a graph-to-graph translation problem, i.e., converting the target molecule graph into a set of reactant molecule graphs. Researchers first identified the reaction center of the target molecule based on GCN, splitting the target molecule into a combination of synthesis subunits. They then used graph VAE to convert each synthesis subunit into the final reactant subgraph. Experimental results showed that G2G significantly outperformed other template-free benchmark models (such as seq2seq models, transformer models, etc.) in terms of Top-1 accuracy metrics, and was comparable to the most advanced template-based methods, such as conditional graph logic network (GLN) models.
5
De Novo Drug Molecule Design Based on Deep Learning
In recent years, DL methods have gained increasing attention in the field of de novo drug molecule design, partly addressing issues such as combinatorial explosion and multi-objective optimization faced by traditional methods. Numerous studies have demonstrated the feasibility of DL methods in de novo drug molecule design, and recent applications of DL in this area have been summarized[29-31]. In this article, I will further introduce the latest research progress. Born et al.[32] constructed a hybrid VAE model to generate candidate molecules with anticancer drug properties. Notably, they not only used molecular SMILES as input but also incorporated disease-related gene expression data for the first time, utilizing anticancer drug sensitivity prediction models as reward functions. The hybrid VAE model consists of two parallel VAEs, one for receiving small molecule SMILES to learn its syntactic rules, and the other VAE for receiving gene expression data to learn its feature representation, then inputting the outputs of these two VAE encoders into the same decoder to generate new molecules, ultimately predicting the activity values of generated molecules against target cells using anticancer drug sensitivity prediction models. Applications in four different cancer types demonstrated that this model can generate molecules with strong inhibitory effects tailored to specific diseases, and the generated molecules are structurally similar to existing drugs in terms of synthesizability and solubility. However, VAEs also have limitations, as they tend to “mimic” training data to the maximum extent, generating molecules structurally similar to training data, resulting in low novelty of generated molecules.
AAE adds a discriminative model to the VAE framework to distinguish between sampled molecules and real samples, training generative and discriminative models based on adversarial ideas, thereby expanding the molecular generation space and addressing the limitations of VAE regarding the novelty of generated molecules. Polykovskiy et al.[33] constructed a new AAE model, conditional AAE, which can generate corresponding molecules based on specified conditions (such as drug molecule target specificity, solubility, and synthesizability). This model constructs encoders and decoders based on long short-term memory networks (LSTM) while employing multi-layer FNNs as discriminative models to determine whether sampled molecules conform to real data distributions and possess desired physicochemical properties, optimizing the model using semi-supervised learning methods. They successfully identified a new Janus kinase 3 (JAK3) inhibitor using this model.
Bagal et al.[34] drew inspiration from the groundbreaking progress of generative pre-training transformer models (GPT) in text generation tasks to construct a new generative model, MolGPT, capable of generating molecules with desired scaffolds and ideal characteristics based on given conditions (input SMILES strings, logP values, synthesizability scores, and topological polar surface area, etc.). MolGPT consists of multiple stacked decoder modules, each containing a masked self-attention layer and multiple fully connected networks, capturing long-distance dependencies between characters in SMILES strings. Compared to other DL models such as VAE and AAE, MolGPT performed better in terms of the effectiveness, uniqueness, and novelty of generated molecules, scoring 0.981, 0.998, and 1.0 respectively.
Goel et al.[35] combined RNN and reinforcement learning to propose a molecular generation model, MoleGuLAR, which can perform multi-objective optimization in terms of molecular drug-likeness and binding affinity. In particular, they introduced a new alternating reward strategy, dynamically changing the reward function during the generation of different molecules, allowing the model to alternately explore different chemical spaces and sample more reasonable molecules. Unlike most previous DL models that can only generate one-dimensional or two-dimensional molecules, Li et al.[36] combined DL with structure-based de novo drug design strategies to develop a new de novo molecular generation model, DeepLigBuilder, capable of directly generating three-dimensional structures of drug-like molecules with high binding affinity. DeepLigBuilder first utilizes a ligand neural network (L-Net) as a graph generation model to generate three-dimensional structures of drug-like molecules, then incorporates target structure information into the model using Monte Carlo tree search methods to search and optimize the binding conformations of molecules at target active sites, ultimately yielding new molecules with high binding affinity. By applying this model to the de novo design of SARS-CoV-2 inhibitors, they identified three novel potential inhibitors with high predicted binding affinity and structures similar to known inhibitors, demonstrating the practicality of DeepLigBuilder in de novo drug design and lead optimization.
To address issues with DL performance on small-scale training datasets, Krishnan et al.[37] designed a generative model based on RNN and a transfer learning-based de novo design process, generating molecules that possess desired drug-like characteristics as well as target specificity. They first pre-trained an RNN generative model using active molecule SMILES data from the ChEMBL database to learn SMILES syntactic rules; then, they docked to obtain molecules with target specificity and performed transfer learning to generate molecules acting on specific targets; additionally, they established another RNN-based predictive model to serve as a reward function evaluating the binding affinity of generated molecules with targets. Moreover, Moret et al.[38] combined RNN generative models with data augmentation, temperature sampling, and transfer learning optimization methods, also achieving the generation of new molecules with desired characteristics under conditions of limited data.
6
ADMET Prediction Based on Deep Learning
Research on the ADMET properties of drugs is also crucial for drug development. Statistics indicate that nearly 50% of candidate drugs fail in clinical trials due to non-compliance with ADMET properties. Therefore, researchers should predict and evaluate the ADMET properties of drug molecules in the early stages of drug discovery and design to reduce the risk of subsequent clinical trial failures. Compared to time-consuming experimental methods, precise and reliable ADMET prediction methods can significantly shorten time expenditures, reduce experimental costs, and improve the efficiency of candidate drug screening. DL-based ADMET prediction methods have emerged as important means for predicting drug ADMET properties.
In recent years, the use of DL methods for predicting small molecule properties has become quite common, with GNN-based methods receiving widespread recognition in academia, yielding more reliable predictive results than other DL methods. In 2018, Wu et al.[39] constructed a DL framework for predicting molecular properties based on the DeepChem platform, termed MoleculeNet. Through this framework, they provided a benchmark for peers to compare the effects and reliability of various models. The framework covers different dataset splitting methods, including scaffold-based and random splits; various feature construction methods, such as ECFP and graph structures; and different network models, such as GCN, MPNN, weave, random forest (RF), and kernel ridge regression (KRR). They trained and tested on various ADMET property-related databases (such as QM8, Clintox, Lipophilicity, BBBP, etc.) through a series of benchmark tests, finding that when applying quantum mechanical properties, physicochemical properties, and physiological properties-related datasets, the best GNN model outperformed the best traditional model. For instance, when training a model using the QM8 dataset to predict the quantum mechanical properties of small molecules, the best-performing traditional model was the KRR model, which achieved a mean absolute error (MAE) of 0.015. In contrast, the best-performing GNN model, the MPNN model, yielded a MAE of 0.0143, which was lower than the KRR model’s test result. Subsequently, researchers established a series of distinctive GNN models from different perspectives. Feinberg et al.[40] constructed a novel GNN model, PotentialNet, whose core idea is to consider distance factors during the update of atomic states, providing a better description of drug molecular structures than commonly used adjacency matrices. This method outperformed traditional machine learning methods and some common GNN models in performance, tested on the QM8 dataset, achieving a MAE of 0.0139 for MPNN, while PotentialNet showed significant improvement, with a MAE around 0.0118. In subsequent research, they further improved upon the PotentialNet model, designing a multi-task PotentialNet model that simultaneously trained on 31 ADMET properties, ultimately predicting all 31 properties[41], such as hERG inhibition, human liver cell clearance rates, half-lives, lipophilicity, etc., comparing it with the RF model. For the majority of properties, the multi-task PotentialNet model exhibited improvements in correlation coefficients (R2) compared to the RF model, such as when splitting datasets using a time-series splitting method, the multi-task PotentialNet model averaged over 64% higher R2 in predicting 31 properties compared to the RF model.
Yang et al.[42] developed a directed message passing neural network (D-MPNN), differing from conventional GNN model practices, considering the bonds between atoms as directed edges rather than conventional undirected edges, updating atomic states based on the direction of edges, thereby reducing ineffective and redundant atomic state updates. Predictive results indicated that D-MPNN outperformed RF models, FNN models, and others across all datasets; for instance, in predicting blood-brain barrier permeability, the D-MPNN model achieved a ROC-AUC of 0.925, while RF and FNN models were only 0.788 and 0.899, respectively. Li et al.[43] proposed a TrimNet model based on a multi-head triadic attention mechanism, analyzing the influence of surrounding atoms on the current atom by providing an adjacency matrix, edge feature matrix, and node feature matrix, thereby efficiently learning latent information from the drug molecular structure represented as a graph, significantly reducing the number of model parameters and computational costs, ultimately achieving good predictive results across multiple datasets, such as a ROC-AUC of 0.948 on the ClinTox dataset.
In addition to GNN-related models, researchers have also attempted other types of DL models and achieved certain results. Kim et al.[44] developed the first interpretable DNN model based on self-attention mechanisms to predict whether drugs exhibit hERG toxicity. Although they only used relatively simple ECFP descriptors and FNN network models, the ROC-AUC on the test set reached 0.893, showing significant improvement over traditional quantitative structure-activity relationship (QSAR) models. Wang et al.[45] constructed a series of derivative networks based on the novel capsule network model (CapsNet), combined with CNN and restricted Boltzmann machines (RBM), achieving a best model ROC-AUC of 0.944. Some research teams have used DL models to directly learn experimental data and predict the pharmacodynamics (PD) and pharmacokinetics (PK) properties of drugs in patients after administration. For example, Lu et al.[46] proposed the Neural-PK/PD model based on RNN models and neural ordinary differential equations (Neural-ODE), innovatively retaining some basic principles of PK/PD in designing the network framework, such as the relationship between the drug’s in vivo effects and dosage and concentration, thus enhancing the predictive accuracy of PK/PD properties.
7
Conclusion and Outlook
DL technology has achieved remarkable predictive capabilities in various stages of drug discovery, changing the drug development process and potentially reducing the costs of drug discovery while improving drug development efficiency. However, existing DL technologies still face numerous challenges. First, most DL technologies heavily rely on substantial computational resources, which somewhat limits the development and application of DL methods. How to reduce the dependence of DL models on computational resources while maintaining predictive accuracy has become a research hotspot in the DL field[47]. One mainstream approach is to prune DL models or improve DL model structures to reduce the number of network parameters and computational loads, thus decreasing the demand for computational resources. Several new lightweight DL models have already been developed and applied[14], such as SqueezeNet, ThiNet, and ShuffleNet. Secondly, the uneven quantity, source, and quality of data samples also limit the establishment and optimization of DL technologies. Training DL models relies on large-scale and high-quality data samples. Effectively conducting few-shot learning is an essential future direction for DL[48], with some methods targeting few-shot learning already in place, such as data augmentation techniques, transfer learning, and multi-task learning strategies. At the same time, the quality of datasets also determines the predictive performance of DL models. The extraction of original data related to drug development and feature construction methods still have shortcomings, impacting the development of high-quality DL models. In recent years, the development of graph neural networks has gradually utilized graphs containing more structural information to represent molecules and apply them in drug discovery, achieving some research progress. Furthermore, hyperparameter search within DL models and the lack of interpretability in internal mechanisms also hinder the development of this technology. In summary, the various shortcomings and challenges faced by DL technologies highlight the need for more researchers from diverse backgrounds to join this field, proposing more sophisticated DL algorithms, and fully combining traditional drug design methods to gradually address the specific issues in various stages of drug development, thereby assisting in innovative drug discovery and further promoting the drug development field towards the intelligent era.
References:
[1] Sengupta S, Basak S, Saikia P, et al. A review of deep learning with special emphasis on architectures, applications and recent trends[J/ OL]. Knowl-Based Syst, 2020, 194: 105596[2021-10-01]. https://doi. org/10.1016/j.knosys.2020.105596.
[2] Fawaz H I, Forestier G, Weber J, et al. Deep learning for time series classification: a review[J]. Data Min Knowl Discov, 2019, 33(4): 917-963.
[3] Segler M H S, Preuss M, Waller M P. Planning chemical syntheses with deep neural networks and symbolic AI[J]. Nature, 2018, 555(7698): 604-610.
[4] Coley C W, Thomas D A, Lummiss J A M, et al. A robotic platform for flw synthesis of organic compounds informed by AI planning[J/ OL]. Science, 2019, 365(6453): eaax1566[2021-10-01]. https:// doi/10.1126/science.aax1566.
[5] Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold[J]. Nature, 2021, 596(7873): 583-589.
[6] Baek M, Dimaio F, Anishchenko I, et al. Accurate prediction of protein structures and interactions using a 3-track network[J]. Science, 2021, 373(6557): 871-876.
[7] Bohr H, Bohr J, Brunak S, et al. A novel approach to prediction of the 3-dimensional structures of protein backbones by neural networks[J]. FEBS Lett, 1990, 261(1): 43-46.
[8] Fariselli P, Olmea O, Valencia A, et al. Prediction of contact maps with neural networks and correlated mutations[J]. Protein Eng, 2001, 14(11): 835-843.
[9] Rahman J, Newton M A H, Islam M K B, et al. Enhancing protein inter-residue real distance prediction by scrutinising deep learning models[J/OL]. Sci Rep, 2022, 12(1): 787[2021-10-01]. https:// doi/10.1038/s41598-021-04441-y.
[10] Yang H, Wang M H, Yu Z H, et al. GANcon: Protein contact map prediction with deep generative adversarial network[J/OL]. IEEE Access, 2020, 8: 80899-80907[2021-10-01]. https://doi/10.1109/ ACCESS.2020.2991605.
[11] Öztürk H, Özgür A, Ozkirimli E. DeepDTA: deep drug-target binding affinity prediction[J/OL]. Bioinformatics, 2018, 34(17): i821-i829[2021-10-01]. https://doi/10.1093/bioinformatics/bty593.
[12] Karimi M, Wu D, Wang Z Y, et al. DeepAffiity: interpretable deep learning of compound protein affiity through unifid recurrent and convolutional neural networks[J]. Bioinformatics, 2019, 35(18): 3329-3338.
[13] Li Y J, Rezaei M A, Li C L, et al. DeepAtom: a framework for protein-ligand binding affiity prediction[J/OL]. 2019[2021-10-01]. https://doi/10.1109/BIBM47256.2019.8982964.
[14] Cheng J, Wang P S, Li G, et al. Recent advances in efficient computation of deep convolutional neural networks[J]. Front Inform Technol Electro, 2018, 19(1): 64-77.
[15] Zheng S, Li Y, Chen S, et al. Predicting drug–protein interaction using quasi-visual question answering system[J]. Nat Mach Intell, 2020, 2: 134-140[2021-10-01]. https://doi.org/10.1038/s42256-020-0152-y.
[16] Cho H, Lee E K, Choi I S. Layer-wise relevance propagation of InteractionNet explains protein–ligand interactions at the atom level[J/ OL]. Sci Rep, 2020, 10(1): 21155[2021-10-01]. https://doi/10.1038/ s41598-020-78169-6.
[17] Zeng Y N, Chen X R, Luo Y J, et al. Deep drug-target binding affiity prediction with multiple attention blocks[J]. Brief Bioinform, 2021, 22(5): bbab117[2021-10-01]. https://doi/10.1093/bib/bbab117.
[18] Sajadi S Z, Zare Chahooki M A, Gharaghani S, et al. AutoDTI++: deep unsupervised learning for DTI prediction by autoencoders[J]. BMC Bioinformatics, 2021, 22(1): 204[2021-10-01]. https:// doi/10.1186/s12859-021-04127-2.
[19] Zeng X X, Zhu S Y, Liu X R, et al. deepDR: a network-based deep learning approach to in silico drug repositioning[J]. Bioinformatics, 2019, 35(24):5191-5198.
[20] Zeng X X, Zhu S Y, Lu W Q, et al. Target identification among known drugs by deep learning from heterogeneous network[J]. Chem Sci, 2020, 11(7): 1775-1797.
[21] Peng J J, Li J Y, Shang X Q. A learning-based method for drugtarget interaction prediction based on feature representation learning and deep neural network[J/OL]. BMC Bioinformatics, 2020, 21(13): 394[2021-10-01]. https://doi/10.1186/s12859-020-03677-1.
[22] Manoochehri H E, Nourani M. Drug-target interaction prediction using semi-bipartite graph model and deep learning[J/OL]. BMC Bioinformatics, 2020, 21(4): 248[2021-10-01]. https://doi/10.1186/ s12859-020-3518-6.
[23] Huang K X, Xiao C, Glass L M, et al. SkipGNN: predicting molecular interactions with skip-graph networks[J/OL]. Sci Rep, 2020, 10(1): 21092[2021-10-01]. https://doi/10.1038/s41598-02077766-9.
[24] Liu Y D, Yang Q, Li Y, et al. Application of machine learning in organic chemistry[J]. Chin J Org Chem, 2020, 40(11): 3812-3827.
[25] Liu B, Ramsundar B, Kawthekar P, et al. Retrosynthetic reaction prediction using neural sequence-to-sequence models[J]. ACS Cent Sci, 2017, 3(10): 1103-1113.
[26] Zheng S J, Rao J H, Zhang Z Y, et al. Predicting retrosynthetic reactions using self-corrected transformer neural networks[J]. J Chem Inf Model, 2020, 60(1): 47-55.
[27] Guo Z L, Wu S, Ohno M, et al. Bayesian algorithm for retrosynthesis[J]. J Chem Inf Model, 2020, 60(10): 4474-4486.
[28] Shi C, Xu M, Guo H, et al. A graph to graphs framework for retrosynthesis prediction[EB/OL]. (2020-03-28)[2021-10-01]. https:// arxiv.org/abs/2003.12725v1.
[29] Liang Li, Deng Chenglong, Zhang Yanmin, et al. Application and challenges of artificial intelligence in drug discovery[J]. Progress in Pharmacy, 2020, 44(1): 18-27.
[30] Xu Y J, Lin K J, Wang S W, et al. Deep learning for molecular generation[J]. Fut Med Chem, 2019, 11(6): 567-597.
[31] Wang M Y, Wang Z, Sun H Y, et al. Deep learning approaches for de novo drug design: an overview[J]. Curr Opin Struct Biol, 2021, 72: 135-144[2021-10-01]. https://doi/10.1016/j.sbi.2021.10.001.
[32] Born J, Manica M, Oskooei A, et al. PaccMannRL: de novo generation of hit-like anticancer molecules from transcriptomic data via reinforcement learning[J]. iScience, 2021, 24(4): 102269[202110-01-01]. https://doi/10.1016/j.isci.2021.102269.
[33] Polykovskiy D, Zhebrak A, Vetrov D, et al. Entangled conditional adversarial autoencoder for de novo drug discovery[J]. Mol Pharm, 2018, 15(10): 4398-4405.
[34] Bagal V, Aggarwal R, Vinod P K, et al. MolGPT: molecular generation using a transformer-decoder model[J]. J Chem Inf Model, 2021[2021-10-01]. https://doi/10.1021/acs.jcim.1c00600.
[35] Goel M, Raghunathan S, Laghuvarapu S, et al. MoleGuLAR: molecule generation using reinforcement learning with alternating rewards[J]. J Chem Inf Model, 2021, 61(12): 5815-5826.
[36] Li Y B, Pei J F, Lai L H. Structure-based de novo drug design using 3D deep generative models[J]. Chem Sci, 2021, 12(41): 1366413675.
[37] Krishnan S R, Bung N, Bulusu G, et al. Accelerating de novo drug design against novel proteins using deep learning[J]. J Chem Inf Model, 2021, 61(2): 621-630.
[38] Moret M, Friedrich L, Grisoni F, et al. Generative molecular design in low data regimes[J]. Nat Mach Intell, 2020, 2(3): 171-180.
[39] Wu Z Q, Ramsundar B, Feinberg E N, et al. MoleculeNet: a benchmark for molecular machine learning[J]. Chem Sci, 2018, 9(2): 513-530.
[40] Feinberg E N, Sur D, Wu Z Q, et al. PotentialNet for molecular property prediction[J]. ACS Cent Sci, 2018, 4(11): 1520-1530.
[41] Feinberg E N, Joshi E, Pande V S, et al. Improvement in ADMET prediction with multitask deep featurization[J]. J Med Chem, 2020, 63(16): 8835-8848.
[42] Yang K, Swanson K, Jin W G, et al. Analyzing learned molecular representations for property prediction[J]. J Chem Inf Model , 2019, 59(8): 3370 -3388.
[43] Li P Y, Li Y Q, Hsieh C Y, et al. TrimNet: learning molecular representation from triplet messages for biomedicine[J]. Brief Bioinform, 2021, 22(4): bbaa266[2021-10-01]. https://doi/10.1093/ bib/bbaa266.
[44] Kim H, Nam H. hERG-Att: Self-attention-based deep neural network for predicting hERG blockers[J]. Comput Biol Chem, 2020, 87: 107286[2021-10-01]. https://doi/10.1016/j.compbiolchem. 2020.107286.
[45] Wang Y W, Huang L, Jiang S W, et al. Capsule networks showed excellent performance in the classification of herg blockers/ nonblockers[J]. Front Pharmacol, 2020, 10: 1631[2021-10-01]. https://doi/10.3389/fphar.2019.01631.
[46] Lu J, Bender B, Jin J Y, et al. Deep learning prediction of patient response time course from early data via neural-pharmacokinetic/ pharmacodynamic modelling[J]. Nat Mach Intell, 2021, 3(8): 696704.
[47] Wu J X, Gao B B, Wei X W, et al. Resource-constrained deep learning: challenges and practices[J]. Sci China Chem, 2018, 48(5): 501-510.
[48] Wang Y, Yao Q, Kwok J T, et al. Generalizing from a few examples: a survey on few-shot learning[J]. ACM Comput Surv, 2020, 53(3): 1-34.
Title Edited: Zhang Xueyuan
Layout Design: Han Hangli

Thank you for reading the original article on the WeChat platform of “Progress in Pharmacy”. Readers are also welcome to reprint and cite. This article is selected from the first issue of “Progress in Pharmacy” 2022.

“Progress in Pharmacy” magazine is co-sponsored by China Pharmaceutical University and the Chinese Pharmaceutical Association, supervised by the Ministry of Education, published monthly, 80 pages, full-color printing. The publication aims to reflect new methods, new results, new progress, and new trends in the field of pharmaceutical research, characterized by reviews, commentaries, and industry development reports, focusing on the progress of pharmaceutical disciplines, technological advancements, and technical information for new drug research and development, making it a professional media focused on the forefront of pharmaceutical technology and industry dynamics.
“Progress in Pharmacy” emphasizes content planning, strengthens manuscript solicitation, deeply excavates and analyzes pharmaceutical information resources, and has begun to take shape in areas such as pharmaceutical discipline progress, research ideas and methods, target mechanism discussions, new drug development reports, clinical medication analysis, and international pharmaceutical frontiers; particularly, the pharmaceutical information content aligns with scientific frontiers and national strategic needs, highlighting foresight, authority, timeliness, novelty, systematization, and practicality. According to the latest statistical data, the average download rate of articles in the publication has ranked first among Chinese pharmaceutical journals for three consecutive years, with a compound impact factor of 0.760, indicating high influence.
The editorial board of “Progress in Pharmacy” is headed by Academician Chen Kaixian, the chief designer of the national major special chemical drugs, with more than a hundred influential experts from government regulatory departments, research institutions, pharmaceutical companies, clinical hospitals, CROs, and financial capital and intellectual property-related organizations.
Contact “Progress in Pharmacy”↓↓↓
Editorial Office Website: pps.cpu.edu.cn;
Email: [email protected];
Phone: 025-83271227.
Welcome to submit and subscribe!
Previous Recommendations
Focusing on the 2021 Nanjing International New Medicine and Life Health Industry Innovation Investment Summit
-
High-energy Review (1) | Based on the forefront of the times, outlining the blueprint for Nanjing’s new medicine and life health industry!
-
High-energy Review (2) | Based on the forefront of the times, outlining the blueprint for Nanjing’s new medicine and life health industry!
-
Starry Night! The complete list of the “2021 China Biopharmaceutical Industry Chain Innovation and Wind and Cloud List” has been announced!
-
New Mechanism of Integration of Industry, Academia, Research, Medicine, and Finance | The founding conference of the “China Biopharmaceutical Industry Chain Innovation and Transformation Alliance” was grandly held in Nanjing!
-
Gathering Talents and Working Together | Directly hitting the scene of the “First China Biopharmaceutical Industry Chain Innovation and Transformation Summit Forum”!
-
Shocking Opening! Experts Gather to Discuss the Frontiers of Gene and Cell Therapy Development
-
Under Capital Transformation, Where is the Path of Innovative Technology in China’s Biopharmaceutical Industry?
-
Strong line-up, full perspective, promoting cooperation! The third session of the “China New Drug Clinical Trial PI Salon” was grandly opened!
-
Summit Focus: Developing the Traditional Chinese Medicine Health Industry to Meet National Strategic Needs
-
Summit Focus: Innovation and Strategic Reserve of Anti-infection Drugs
-
Focusing on Investment Hot Tracks, 12 Projects Shine!
-
When Artificial Intelligence Meets Biomedicine, What Sparks Will Emerge?
-
Grasping New Trends in Drug Innovation, Seizing the New Track of Improved New Drugs
Focusing on the 2020 Nanjing International Life and Health Technology Conference
-
“Iron Shoulders Carry Righteousness, Wonderful Hands Write Articles”—The Sixth Editorial Elite Seminar of “Progress in Pharmacy” was held in Nanjing
-
“China Biopharmaceutical Industry Chain Innovation and Transformation Alliance” Inaugural Ceremony and Preparatory Meeting Record
-
2020 Nanjing International Life and Health Technology Conference grandly opened, marking the beginning of a new era of innovation collaboration in China’s biopharmaceutical industry chain!
Focusing on the 2020 Nanjing International New Medicine and Life Health Industry Innovation Investment Summit
-
Summit Focus: In September, the innovative city once again exerts its strength, with over a hundred industry leaders discussing the new landscape of Nanjing’s life and health industry!
-
Summit Focus: In the post-epidemic era, how can new drug R&D in China go against the trend and make proactive layouts?
-
Summit Focus: Exploring the collaborative innovation path of small molecule innovative drugs and high-end formulations between industry, academia, and research
Pharmaceutical Microvision
-
Pharmaceutical Microvision | Academician Chen Kaixian: The development of China’s biopharmaceutical industry and the pace of new drug R&D are accelerating, with both opportunities and challenges in the future.
-
Pharmaceutical Microvision | Academician Shen Hongbing of Nanjing Medical University: The integration and innovation of medicine and pharmacy promote the development of Nanjing’s new medicine industry.
-
Pharmaceutical Microvision | Academician Deng Zixin: China’s new drugs are catching up, and there is no end to innovation.
Pharmaceutical Interviews
-
Pharmaceutical Interview | Professor Zhu Xun: How can Nanjing’s biopharmaceutical industry leverage its latecomer advantages? Rather than filling gaps, focus on strengths!
-
Pharmaceutical Interview | Dr. Zhang Lianshan: Building landmarks for Nanjing’s new medicine and life health industry, strengthening the aggregation of industry elements is key.
-
Pharmaceutical Interview | Huang Bin from AstraZeneca: Relying on AstraZeneca’s global network, creating the preferred entry point for innovative enterprises to develop the Chinese market and connect with the global market.
I know you’re watching!
