Paper Overview
Title
Research Progress of Machine Learning in Protein Function Prediction
Authors
Chi Yanfei, Li Chun, Feng Xudong
Citation
Chi Yanfei, Li Chun, Feng Xudong. Research Progress of Machine Learning in Protein Function Prediction [J]. Journal of Biotechnology, 2023, 39(6): 2141-2157.
Abstract:Proteins are essential compounds in organic life forms, playing various important roles in life activities. Understanding protein functions aids research in fields such as medicine and drug development. Furthermore, the application of enzymes in green synthesis has attracted much attention. However, due to the diversity of enzyme types and functions, obtaining specific functional enzymes is costly, limiting their further application. Currently, the specific functions of proteins are mainly determined through experimental characterization, a method that is cumbersome and time-consuming. With the rapid development of bioinformatics and sequencing technologies, the number of sequenced protein sequences far exceeds the number of sequences with functional annotations, making efficient prediction of protein functions crucial. With the booming development of computer technology, data-driven machine learning methods have become an effective solution to these challenges. This paper provides an overview of protein functions and their annotation methods, as well as the development and operational processes of machine learning, focusing on the application of machine learning in enzyme function prediction, and proposes prospects for the future development direction of artificial intelligence-assisted efficient research on protein functions.
Proteins are one of the indispensable material foundations of various life activities, exhibiting diversity in their biological functions and playing important roles in various life activities. The vast majority of enzymes also belong to proteins and play significant roles in the field of biomanufacturing[1]. Researching protein functions can assist in exploring the molecular mechanisms of various life activities, facilitating rapid development in medicine and drug research[2-3]. Currently, protein functions are primarily characterized through biochemical experiments; however, this method is time-consuming, labor-intensive, and costly. With the rapid advancements in biotechnology, billions of protein sequences are continuously being discovered, and the number of sequenced protein sequences far exceeds the number of sequences with known functions. Relying solely on experimental verification for protein function annotation can no longer meet current demands[4-5].
As computer science continues to develop, especially during this era of rapid advancements in artificial intelligence, efficiently and accurately predicting and annotating protein functions using computational methods has become a current research hotspot. Therefore, this paper aims to summarize the research progress in the field of artificial intelligence-assisted protein function prediction, primarily introducing existing protein function annotation methods and concepts related to machine learning, with a focus on the practical applications of machine learning methods in enzyme function prediction.
1 Protein Function Annotation
Proteins are an important part of living organisms, being complex in structure and diverse in function, and are significant for various life activities. Proteins participate in important physiological processes, such as cell signaling, catalytic reactions, immune regulation, and substance transport, also providing energy for the metabolism of living organisms[6]. Exploring protein functions can assist in studying the molecular mechanisms of life activities[7] and plays an important role in research and drug development in the medical field[2-3]. Protein function typically focuses on the role of individual protein molecules, such as catalysis for given reactions or molecular binding. This localized function is also referred to as the molecular function of proteins. On the other hand, proteins are also defined as elements in their interaction networks, meaning that proteins play roles within the extended networks of their interacting molecules, which is referred to as contextual function or cellular function[8-9].
With the rapid development of sequencing technologies and genomics, an increasing number of protein sequences are being determined. Accurately annotating the functions of proteins from their sequences can help broaden our understanding of life forms in nature and their activities, significantly promoting the development of drug research, medicine, and biogreen synthesis[10]. Currently, the UniProt database contains over 200 million protein sequences[10], while only about 1% of the protein sequences have undergone functional annotation[11]. For example, UDP-glycosyltransferases (UGTs) are a common type of glycosyltransferase belonging to the GT1 family of proteins, with over 30,000 GT1 members identified, yet only 413 UGTs have had their functions confirmed through experiments[12]. Additionally, over 30% of proteins with resolved structures still lack functional annotations[9]. Therefore, predicting protein functions is currently an important research direction.
Currently, biochemical experiments are the main method for characterizing and annotating protein functions; however, this method is costly and time-consuming, and the gap between the number of proteins with annotated functions and those with unknown functions is widening. With the rapid development of artificial intelligence, many computational methods have been applied to solve the problem of protein function prediction. Compared to time-consuming experimental verification methods, computational methods can predict the functions of large numbers of proteins simultaneously. Predicting and annotating protein functions using artificial intelligence is one of the current research hotspots, and efficiently and accurately predicting various protein functions can greatly advance research in drug target discovery, biological activity mechanisms, and biosynthesis.
2 Traditional Protein Function Prediction Methods
2.1 Sequence-Based Methods
Sequence-based methods primarily approach from an evolutionary perspective, as the structure of a protein determines its function, which in turn is determined by its amino acid sequence. Homologous proteins have sequences that exhibit a certain degree of similarity, thus sharing similar functions[13]. Therefore, sequence-based methods mainly determine the homology between proteins by comparing the similarity between sequences. The most widely used comparison tools currently are BLAST[14] and HMMER[15]. BLAST primarily estimates the function based on the similarity of the target protein to known functional proteins, serving as an analysis tool for comparing similarity between various protein databases or gene databases, mainly comprising the Needleman-Wunsch algorithm and the Smith-Waterman algorithm[14]. HMMER primarily employs hidden Markov models to annotate target proteins, achieving higher accuracy in distant protein annotations compared to BLAST[16]. For functional predictions of multiple genomes or protein sequences, after HMMER annotation, tools such as ClustalW[17] can be used for multiple sequence alignment and constructing phylogenetic trees to explore evolutionary relationships between sequences, thereby inferring sequence homology and functional similarity. Currently, sequence-based methods have been widely applied in the field of protein function annotation and have achieved practical applications in many studies.
Additionally, proteins typically consist of at least one conserved structural domain, with shorter and conserved segments within the domain referred to as motifs, which are often associated with specific biological functions. Motif-based methods have been successfully applied to protein recognition and classification. For example, a conserved sequence of 44 amino acids at the C-terminal of UDP-glycosyltransferases from plant sources serves as a binding site for glycosyltransferases and sugar donors, known as the plant secondary product glycosyltransferase motif (PSPG box) conserved region[18], which is regarded as a key feature when mining and screening UDP-glycosyltransferases in plant genomes.
However, inferring protein functions based on sequence homology typically requires a similarity greater than 60% between sequences for the method to achieve a certain level of credibility[14]. Moreover, there is no absolute correlation between homology and protein function; two sequences being homologous only indicates they share a common ancestor, but they may not be functionally similar[19]. For example, the similarity of enzyme sequences with functional annotations does not show a significant correlation with their substrate specificity, which is detrimental to direct functional predictions and applications of enzymes. For instance, UDP-glycosyltransferases primarily utilize uridine diphosphate-sugar (UDP-sugar) as a sugar donor. The UDP-glycosyltransferases VvGT5 and VvGT6 from grapes have 91% sequence similarity, yet VvGT6 can use UDP-Glc and UDP-Gal as sugar donors, while VvGT5 can only use UDP-GlcA as a donor[20]. Similarly, UGT73P12 from Ural licorice and GmSGT2 (UGT73P2) from soybeans have 75% sequence similarity, but GmSGT2 uses UDP-Gal as a sugar donor to transfer galactosyl to soybean saponin B-3-O-monoglucuronic acid to synthesize soybean saponin III, while UGT73P12 transfers the glucuronic acid moiety of UDP-GlcA to glycyrrhizic acid 3-O-monoglucuronic acid to synthesize glycyrrhizic acid[21]. Similarly, low similarity sequences may also exhibit similar functions; for example, GuGT33 (UGT84F6) and GuGT37 (UGT71S4) from light-fruited licorice have similar substrate specificities, yet their protein sequence similarity is only 23.7%, and they belong to different UGT families[22-23]. These results indicate that it is insufficient to accurately determine the biochemical functional characteristics of UDP-glycosyltransferases based solely on sequence similarity; a substantial amount of experimental validation is still required to ultimately determine enzyme functional characteristics after phylogenetic analysis based on homology. Therefore, while sequence homology methods have been widely used, many issues still need to be addressed.
2.2 Structure-Based Methods
Based on the theoretical foundation that protein structure directly determines its biological function, proteins with similar spatial structures often share the same function[13]. Structure-based protein function prediction methods mainly achieve this through two approaches[16]: global folding similarity comparison of protein structures and local active site feature description. The three-dimensional spatial structure of proteins is evolutionarily more conserved than the one-dimensional amino acid sequence[24]. Currently, most studies primarily infer the functions of unknown proteins based on the similarity of specific binding regions in their three-dimensional structures. For example, the binding site of UDP-glycosyltransferases is a narrow pocket between the C-terminus and N-terminus structural domains[25]. The function of enzymes is almost entirely determined by these active sites on the protein structure, as residues surrounding the active sites of proteins maintain high conservation throughout evolution[13]. Many tools have been proposed to predict protein functions by comparing unknown proteins with structures in the protein data bank (PDB)[26]. For example, the online protein structure comparison website FATCAT[27] (www.fatcat.burnham.org), the well-performing structure comparison tool DeepAlign[28], and the protein structure classification database CATH-Gene3D[29] (www.cathdb.info) are among them. FATCAT can compare the target protein with structures in the PDB database for similarity and structural alignment; DeepAlign not only compares the rigid structures of proteins but also considers the impact of evolutionary relationships and hydrogen bond similarities on structural differences between proteins; CATH classifies proteins in the PDB database based on specific spatial structural information. Although structure-based methods for predicting functions have yielded a wealth of research results, many shortcomings still exist. This method requires a large amount of known functional protein structural data for modeling and relies heavily on the accuracy of the models[13]. However, protein structure resolution has always been a complex and challenging scientific task in the life sciences, and the amount of protein data with resolved structures is still insufficient to build an adequately accurate model. For instance, for UDP-glycosyltransferases, only 51 crystal structures have been resolved[12,30-31]. Although the number of proteins with resolved structures continues to increase with advancements in protein structure resolution technologies, making structure-based prediction methods gradually more reliable, there remains a long way to go.
3 Machine Learning-Based Protein Function Prediction Methods
Intelligence is a characteristic related to humans, and learning is the most significant intelligent behavior of humans. Machine learning, as an important branch of artificial intelligence, integrates theories from various disciplines such as statistics and probability theory. Its main goal is to enable machines to learn and mimic human thinking patterns, automatically adjusting the weights of input factors, learning comprehensively, and quickly outputting results. Deep learning, a subfield of machine learning, represents the closest approach to human brain intelligence in learning and cognition methods, achieving significant progress in speech and image recognition and transformation across various fields, with enormous application and commercial potential.
Machine learning was initially established on the research of artificial neural networks. In 1989, Lecun et al.[32] proposed the first successfully trained artificial neural network computational model: the convolutional neural network (CNN) computational model, which is widely used across various fields of computer technology today. The basic structure of CNN (Figure 1) mainly consists of an input layer, convolutional layers (activation functions), pooling layers, fully connected layers, and an output layer. The convolutional layer is a unique structure of CNN, serving to perform convolution operations on input data to extract feature information. Activation functions are nonlinear functions with properties such as continuity and differentiability; the most commonly used activation functions include Relu, Tanh, and Sigmoid. Pooling layers operate between consecutive convolutional layers to reduce dimensionality and prevent excessive data and parameter volumes. The fully connected layer appears in the last layer of the network, primarily connecting all data matrices, combining all local features into global features, and ultimately outputting data.
By the end of the 20th century, the explosive growth of data, continuously improving and optimizing computer technologies and algorithms led to the official arrival of the artificial intelligence boom[33]. Since then, artificial intelligence technologies, represented by machine learning, have begun to thrive in daily human life, such as in gaming, facial recognition, speech recognition, autonomous driving, and medical imaging analysis[34]. Currently, machine learning has achieved abundant research and application results in both academic and industrial fields, promoting rapid development in scientific research and practical applications, with artificial intelligence becoming one of the hottest research directions. For instance, after DeepMind developed the deep learning Go program AlphaGo, which defeated the human world champion, it began to venture into the life sciences, developing a protein three-dimensional structure prediction program based on deep neural network algorithms called AlphaFold. This program can predict the three-dimensional structure of a protein by inputting an unknown protein sequence; in December 2020, the improved AlphaFold2[35-36] achieved an accuracy level nearly matching experimental precision when compared with established experimental techniques such as nuclear magnetic resonance, X-ray crystallography, and cryo-electron microscopy, with predicted results differing from experimental measurements by only one atom. With the rapid development of bioinformatics, biological data is gradually accumulating, and an increasing number of researchers in the life sciences are using machine learning to assist research, thus promoting faster and more efficient development of the discipline. Currently, machine learning in the life sciences is not only applied to predicting unknown protein structures and functions but also to genomic research and drug development, assisting in drug target research[37] and predicting drug toxicity[38]; in medical imaging, machine learning can also be used to identify lesions based on big data to assist diagnosis[39].
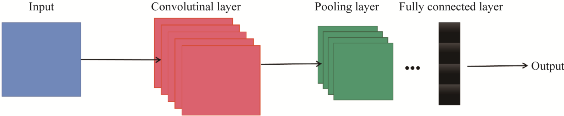 Figure 1 CNN Basic Structure
Figure 1 CNN Basic Structure
Currently, machine learning-assisted life sciences have achieved fruitful research results and practical applications. With the continuous development of computer technology, it will undoubtedly promote faster and more efficient development in this field.
3.1 Classification and Algorithms of Machine Learning
Currently, protein function prediction is primarily a classification problem, mapping sequences and their feature information as inputs to discrete functional information outputs. The machine learning algorithms that can be used are divided into two categories: supervised learning and reinforcement learning. Supervised learning is further divided into three types based on the presence or absence of data labels: traditional supervised learning, unsupervised learning, and semi-supervised learning.
Traditional supervised learning datasets mainly consist of initial training data and manually annotated training targets. This step is the most time-consuming yet crucial in the entire workflow, as researchers hope that the computer can summarize specific object classification rules from training data based on the annotated features using specific algorithms, which can then be applied to predict testing data, ultimately outputting learning results with specific labels. Traditional supervised learning includes classic algorithms such as support vector machines (SVM), artificial neural networks (neural networks), and deep neural networks (deep neural networks); typical applications include regression analysis and task classification. Unsupervised learning is primarily applied when training data has not been manually classified and labeled, allowing the computer algorithms to autonomously learn and summarize patterns between data based on similarity principles, ultimately obtaining the features of the training data. Typical algorithms for unsupervised learning include clustering, expectation-maximization algorithm (EM), and principal component analysis (PCA); typical applications include clustering and anomaly detection. Semi-supervised learning lies between the two, where only a portion of the training data is labeled. Currently, the majority of software used in everyday internet life employs semi-supervised learning algorithms. Semi-supervised learning mainly addresses the problem of reduced model performance in traditional supervised learning when there are insufficient labeled samples by introducing unlabeled samples into model training. In practical applications, obtaining labeled samples is often costly, while unlabeled samples can be easily collected in large quantities. Therefore, in practical applications, when the amount of labeled samples is too small, semi-supervised learning algorithms can achieve comparable or even better results than traditional supervised learning trained with a large number of labeled samples, significantly improving production efficiency. Over the past few decades, an increasing number of machine learning algorithms have been continuously updated and developed for application in various fields.
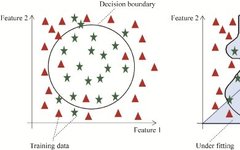
However, there are corresponding difficulties in the practical application of machine learning (Figure 2), such as overfitting and underfitting due to a small number of feature samples, which are challenges that machine learning methods face in the field of protein function prediction. Underfitting refers to a situation where the model uses too few parameters, making it difficult to fit the training data, resulting in poor performance on the training set, validation set, and test set. Overfitting occurs when too many parameters are used, leading to excessive fitting of the model to the training data, meaning the model performs well on the training set but poorly on the validation and test phases. Model construction usually requires selecting and testing multiple algorithms based on practical purposes, thus, currently, there is a lack of a universal machine learning model in the field of protein function prediction.
3.2 Common Protein Databases
Machine learning algorithms depend on large amounts of data, thus the quantity and quality of the training datasets are crucial. The most frequently used databases in protein engineering are InterPro[41], UniProtKB[42], and PDB[26]. UniProtKB is the largest sequence database, composed of the SwissProt and TrEMBL datasets. SwissProt contains manually annotated and non-redundant data, ensuring a certain quality; TrEMBL includes some functionally unknown protein sequences that still require manual annotation. InterPro integrates multiple protein-related databases, containing over 38,000 entries, and has a protein sequence function annotation tool called interproscan, which UniProtKB mainly utilizes to annotate unknown protein sequences. PDB contains nearly 200,000 high-resolution protein three-dimensional structural information, making it the largest protein 3D structure database. Gene ontology (GO)[43] can systematically annotate the attributes of species genes and their products, and many studies have applied the GO term database to protein function prediction.
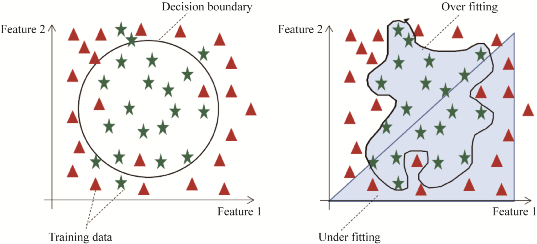 Figure 2 Problems Commonly Encountered in Machine Learning Table 1 Summary of Commonly Used Databases in Protein Function Prediction
Figure 2 Problems Commonly Encountered in Machine Learning Table 1 Summary of Commonly Used Databases in Protein Function Prediction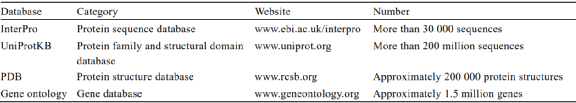
The quantity and quality of data in the above databases are crucial for establishing machine learning prediction models. Among them, the protein sequence database is the most abundant, followed by the protein structure database. However, in practical research on protein function prediction, it is hoped that amino acid sequences can directly reflect the function of the protein. Therefore, for protein sequences, datasets can be organized based on the physical and chemical properties of amino acids. Furthermore, in the prediction of protease functions, due to the complexity and diversity of enzyme reaction types, mechanisms, cofactors, and conditions, predicting their specific functional characteristics is a highly challenging task. Currently, more databases need to be constructed to address this challenge.
3.3 Selection of Protein Function Features
One key step in establishing a machine learning model is identifying suitable features. Due to the richness and diversity of protein functions, using machine learning techniques to predict protein functions requires determining suitable feature labels based on specific needs. Proteins with similar functions usually exhibit similar physicochemical properties in terms of sequence and structure, such as isoelectric point, molecular weight, surface tension, polarity, hydrophobicity, and charge number. Additionally, biological properties such as amino acid features, ligand properties, and structural variability[19] are also considered. Machine learning methods primarily utilize computers to capture the relationships between these feature information and protein functions. Amino acid sequence features include the composition, distribution, and conservation of amino acids[19], which not only significantly impact functional predictions but have also been applied in predicting protein-protein interaction networks[44]. The most notable feature in sequence features is the sequence motif, which exists in the vast majority of proteins. Additionally, the protein-protein interaction (PPI) network is a mathematical representation of the physical contact between proteins[9], playing an important role in elucidating the specific connections between protein functions, and is widely used as a feature in the field of protein function prediction.
Similar to homology comparison methods, machine learning methods for predicting protein functions also require collecting a large amount of data from known functional proteins, learning the relationships between protein features and functions, and establishing computational models to ultimately predict the specific functions of target proteins (Figure 3). However, both sequence homology and structure-based methods depend on sequence similarity or structural similarity results, while machine learning methods can extract protein features from different angles, such as sequences, structures, and protein networks, and map them to corresponding functions, thus improving the issues present in traditional methods based on sequence homology. For example, the protein family database Pfam has grown by 5% in protein sequences over the past few years, yet at least one-third of proteins cannot be functionally annotated by direct comparison with sequences that have known functional characterization[7,45]. Bileschi et al.[46] used a deep learning model (ProtCNN) to predict functions for 17,929 unaligned protein sequences from the Pfam database, showing higher accuracy compared to existing traditional methods when classifying test sequences. Furthermore, they combined this model with traditional alignment methods such as BLAST and HMMER to accurately classify sequences with low similarity to training data, indicating that complementing deep learning with existing methods can effectively improve prediction efficiency and model performance. Machine learning models can predict a large number of unknown functional protein sequences at once, not only achieving better prediction results compared to traditional alignment methods but also greatly increasing prediction speed, saving time. For example, Lv et al.[16] extracted features from protein sequences and protein interaction networks, applying deep neural network models to predict human protein functions, achieving an accuracy rate 51.3% higher than the traditional BLAST method. Additionally, the BLAST method requires a biological background for the unknown protein, while machine learning can infer biological characteristics of unknown protein sequences and predict their functions without physical or biological knowledge.
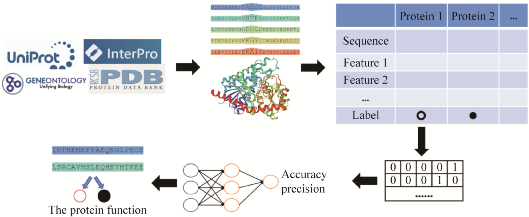 Figure 3 Flowchart of Machine Learning Predicting Protein Functions
Figure 3 Flowchart of Machine Learning Predicting Protein Functions
Currently, predicting protein functions based on machine learning primarily involves analyzing key residues, their physicochemical properties, and conservation levels[19]. For example, Corral-corral et al.[47] explored the relationship between key residues, protein structure, and protein function using various machine learning algorithms and different protein feature representation methods. The final results indicated that using the physicochemical properties of key residues as features yielded the highest accuracy for the machine learning model. Gado et al.[48] utilized the number of residues and amino acid types within loops as training features, employing machine learning methods to accurately distinguish between the CBH and EG subtypes of 1,748 family glycoside hydrolases. In 2018, GT-predict[49] selected 54 enzymes from the glycosyltransferase superfamily 1 from Arabidopsis and 91 substrates for function prediction. The authors systematically combined sequence and substrate molecular physicochemical properties, including clogP, molecular area, and the number/type of nucleophilic groups, as well as structural information, achieving an accuracy rate exceeding 69% in other four glycosyltransferases with known functional characterization from plants and microorganisms. GT-predict highlights the importance of data features in improving model accuracy; however, this prediction tool directly outputs functional information based on sequence alignment with known functional features, without considering the chemical region selectivity issue, leading to overfitting problems. Therefore, machine learning methods highly depend on data quality, and the accuracy of prediction models still needs further enhancement. Additionally, although machine learning has achieved certain research results and application potential in fields such as protein function prediction, there is currently no algorithm that is optimal for all tasks, according to the “No Free Lunch Theorem”[50]. How to quickly obtain more accurate and suitable training data for machine learning algorithms and find and establish more efficient data feature representation methods to improve model accuracy is an urgent issue that needs to be addressed in the application of machine learning across various fields.
4 Application Progress of Machine Learning in Protein Function Prediction
With the development of sequencing technologies, the gap between the number of proteins sequenced and the number of proteins with known functions is increasingly widening. To address this issue, the critical assessment of functional annotation (CAFA) global challenge[51] proposed a delay performance evaluation method, using annotated protein sequences from published articles or databases as training and testing sets, which can enhance the reliability of functional prediction model algorithms. Currently, most algorithms for protein function prediction, including those based on sequence alignment and machine learning methods, refer to CAFA’s delay assessment. At present, both the computer and biochemical engineering fields are experiencing vigorous development, and how to effectively combine the two to utilize computational methods to assist research in the biochemical engineering field has also been a research hotspot. For example, although biocatalytic chemical reactions have advantages such as being environmentally friendly and highly specific, the process of mining and identifying specific functional enzymes is costly. Using computational methods to assist in the efficient mining of enzymes and guiding biochemical experiments will undoubtedly yield significant results and effectively address the substantial gap between the number of unknown functional protein sequences and those with functional annotations.
4.1 Traditional Machine Learning Methods
For different datasets, machine learning predicts protein functions primarily by using protein sequences and structures as inputs to generate features, constructing and optimizing models using different algorithms, such as KNN (k nearest neighbors) algorithm[52], naive Bayes (NB)[53-54], SVM[55], and neural networks[56]. Extracting features solely from sequences is the simplest classification model for protein functions[57]. Although protein structures contain richer information than protein sequences, current research results indicate that directly predicting protein functions from amino acid sequences can also achieve good accuracy[58]. As shown in Table 2, Che et al.[59] collected 59,764 protein sequences from the SwissProt database after removing highly similar sequences, extracting features using the ACC control algorithm, and constructing models using the KNN algorithm for enzyme function prediction. The results indicated that this method achieved a classification accuracy of 94.1% for single-functional enzymes and 91.25% for multifunctional enzymes, and the model has been made into an online prediction website, significantly enhancing its practicality. Osman et al.[60] selected 3,200 types of enzymes from six superfamilies in the PDB database and tested 2,000 enzymes using a neural network model with a hybrid GA-BP algorithm, ultimately achieving an average accuracy of 72.94%. Mohammed et al.[61] collected 64,948 enzyme sequences from the EXPASY enzyme database and 128,292 non-enzyme protein sequences from the SwissProt database, utilizing the Pfam, Superfamily, and Prosite domain databases for feature extraction regarding function, structure, and motif or active site regions, training models using five different machine learning algorithms (KNN, SVM, decision trees, random forests, and naive Bayes), and achieving an accuracy rate between 97% and 99% with their ECemble model, outperforming traditional BLAST and similar open-source methods like EFICAz[71]. Additionally, the authors applied ECemble to the gut metagenome, predicting enzymes encoded by the human gut microbiome, aiding in understanding the role of microbial-encoded enzymes in the health functions of the human metabolic system.
Table 2 Applications of Different Machine Learning Algorithms in Protein Function Prediction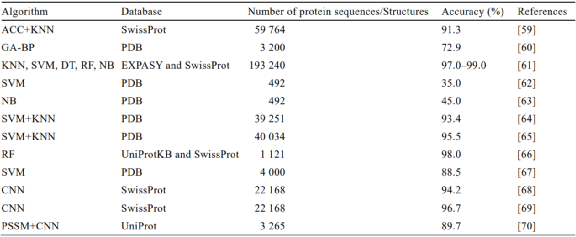
Predicting protein functions based on amino acid sequence features primarily relies on homology principles. However, when a protein lacks a similar protein sequence with a known function, methods that rely solely on sequences become ineffective. In 2005, Dobson and Doig[62] proposed a method to predict protein functions based on structural information. They selected 492 protein structure data from six enzyme classifications based on EC numbers, using simple crystal structure attributes such as secondary structure content and surface properties as feature information. The resulting SVM model achieved an accuracy rate of 35%. In 2006, Borro et al.[63] built on Dobson and Doig’s research, using Bayesian algorithms to improve accuracy to 45%. In 2016, Amidi et al.[64] combined structural information with amino acid sequence alignment, training SVM and KNN algorithms on a dataset of 39,251 protein data from the PDB database, achieving a classification accuracy of 93.4%. In 2017, Amidi et al.[65] combined protein structure and amino acid sequence information, constructing a multi-enzyme function prediction model using SVM and nearest neighbor algorithms, achieving an accuracy rate of 95.5% for predicting the first digit of the EC number in 40,034 enzymes from the PDB database. This result also indicates that combining protein information can provide more accurate predictions in enzyme function prediction.
Using sequence or structure from different data sources as input information results in overly simplistic model input, leading to insufficient accuracy and applicability. In enzyme databases, functional annotation labels are typically added to enzyme sequences for classification and reference. However, due to the diversity of enzyme types and catalytic functions, predicting protease functions using machine learning is not a simple single-label classification problem. In 2014, Nagao et al.[66] first applied the random forest algorithm to predict the fourth digit of EC numbers for each homologous superfamily enzyme, using similarities between active site residues, ligand binding residues, and conserved residues, in addition to full-length sequence similarity as input features. This model created a dataset from the UniProtKB and SwissProt databases, annotating and classifying through the protein structure domain annotation database Gene3D, ultimately achieving an accuracy rate of 98% when tested on 1,121 enzymes from 306 CATH homologous superfamilies. The model also identified some specific residues through random forests, discovering that the number of active residues contained in superfamilies with high functional differentiation is the highest. Integrating various data types about proteins is beneficial for predicting their functions. For example, Srivastava et al.[67] classified 4,000 sequence data from the PDB protein database based on EC numbers and enzyme names, dividing them into training and testing sets. They predicted the EC categories of training set proteins using SVM and random forest algorithms combined with seven features, including primary structure, structural molecular weight, ligand molecular weight, and chain length, resulting in an overall accuracy of 88.49% for the SVM model, outperforming random forests at 53.9%. In practical situations, various biological and physicochemical properties affecting protein functions are not entirely independent, and the contributions of various feature combinations to protein annotation also need to be considered. GOLabeler[72] is a model that uses sequences as input, integrating various feature information such as sequence homology, protein structural domains, motifs, and physicochemical properties of amino acids. It employs five different classifiers (BLAST-kNN, LR-3mer, LR-InterPro, LR-ProFET, and naive GO item frequency calculation) to learn different features and subsequently adjusts weights for each classifier based on the degree of influence of different features through a learning-to-rank algorithm (LTR). In datasets from CAFA1 and CAFA2, GOLabeler significantly outperformed other participating models in predicting GO term labels for unknown functional proteins, achieving the best performance in CAFA3 and CAFA-π challenges.
4.2 Deep Learning Methods
Deep learning can efficiently handle complex and highly nonlinear big data problems compared to traditional machine learning. CNN is a type of deep learning model widely applied across various research fields. It can fully learn the feature relationships of training data through the powerful fitting ability of neural network algorithms, thus outputting prediction results with high accuracy. In protein function prediction, deep learning methods represented by CNN typically achieve better fitting results than traditional machine learning algorithms. For instance, Li et al.[68] developed a novel end-to-end feature extraction and classification deep learning model, DEEPre, constructing a sequence dataset based on the ENZYME database. The model framework inputs raw sequence encoding, extracting convolutional and sequential features based on classification results, directly improving model performance and successfully predicting enzyme functions by predicting EC numbers. DEEPre achieved an accuracy of 94.15% for predicting enzyme subclasses using a validation test set of 22,168 enzyme and non-enzyme sequences collected from the SwissProt database. Building on this, Zou et al.[69] proposed a hierarchical multi-label deep learning enzyme function prediction model, mlDEEPre, which first predicts whether a given unknown protein sequence is a single-functional enzyme or a multifunctional enzyme, further predicting its specific function for multifunctional enzymes, achieving an accuracy of 96.7%.
GO terms[43] can accurately describe various levels of protein functions, aiding in understanding the molecular or biochemical functions of proteins. Currently, an increasing number of deep learning methods utilize GO terms to assist in predicting protein functions[75]. For example, DeepGO[76] is a completely data-driven deep learning model for predicting protein functions, built without relying on any manually input feature label information. Its dataset includes 60,710 protein sequences from the SwissProt database, with 27,760 categories of GO terms, covering over 90% of the annotated protein sequences in SwissProt. DeepGO employs deep learning methods to learn two features useful for predicting protein functions (protein sequences and positions of proteins in interaction networks), demonstrating significant improvements compared to traditional methods such as BLAST, as confirmed by CAFA evaluation standards, and showing good predictive performance in predicting the cellular localization of proteins. DeepGOplus[77] uses data from the CAFA3 challenge as its dataset, combining sequence similarity predictions with CNN models, enabling rapid functional predictions for any protein, annotating 40 proteins per second. Additionally, since DeepGOPlus does not impose restrictions on the length of amino acid sequences, it can be applied for genome-scale annotations of protein functions. Furthermore, selecting appropriate features is crucial for the predictive performance of machine learning models; for instance, compared to DeepGO, which extracts simple 3-mer sequence features, DeepFunc[78] uses more informative sequence features, such as domains, families, and motifs related to protein chains, leading to better predictive performance than DeepGO. Moreover, position-specific scoring matrices (PSSM), primarily used in bioinformatics for assessing homologies between proteins, can provide a new approach to predicting protein functions when combined with deep learning. For example, Le et al.[79] proposed a method for predicting guanosine triphosphate (GTP) binding sites in transport proteins based on PSSM and biochemical characteristics, achieving an accuracy rate of 95.6% for identifying transport protein categories from a molecular function perspective. Additionally, Le et al.[70] collected 682 SNARE proteins and 2,583 non-SNARE protein sequences from the UniProt database, removing redundant sequences with similarities greater than 30% using BLAST, and inputting PSSM into the CNN framework, ultimately achieving an accuracy of 89.7% in identifying SNARE proteins.
5 Summary and Prospects
Through artificial intelligence strategies, particularly machine learning methods for predicting protein functions, significant advancements have been made in the field of protein function prediction in recent years. Machine learning methods can infer biological information of unknown functional proteins directly by learning the mapping relationship between protein sequences and functions without any biological background knowledge. However, the diversity of enzyme types and catalytic mechanisms also poses significant challenges for these methods, as a single machine learning algorithm struggles to cover all functions and address corresponding complex relationship problems. Currently, there is a lack of a universal model capable of predicting the specific functions of all proteins. Similarly, data collection and feature processing require strict quality control, as collecting new data for model testing is often the most labor-intensive and time-consuming aspect of the entire research process, and the data formats are challenging to standardize. Experimental data from different sources exhibit varying reliability, and the differences between data need to be considered. Furthermore, the number of proteins among different families varies greatly; some families may have thousands of annotated sequences, while others may only have a few dozen, which is not conducive to model training from a machine learning perspective. Families with small data volumes are referred to as low-sample-label[80] in the computer field, and accurately annotating low-sample-label proteins is a challenge in the field of protein function prediction research. Therefore, improving data quality and feature representation to obtain more accurate computational models is a key issue to be addressed when using machine learning to predict protein functions.
Moreover, natural language processing (NLP) is also an important branch of artificial intelligence, involving the processing, understanding, and application of human language by computers to perform target tasks. Protein sequences are inherently similar to language, where letters form words and sentences with specific meanings; similarly, various arrangements of amino acids can form proteins with diverse structures and functions. Therefore, in recent years, NLP technologies have attracted attention in protein research and design[81], such as ProGen[82] and ProtGPT2[83], which generate protein sequences with specific functional characteristics and energies close to natural structures. In the future, NLP technologies are expected to be more widely applied in the field of protein function prediction, aiding in further research on the relationships between protein sequences, structures, and functions.
 Feng Xudong
Feng Xudong
Beijing Institute of TechnologySpecial Researcher,Doctoral Supervisor. He obtained his Ph.D. in Engineering from the University of Auckland (New Zealand) in 2014 and is currently the Deputy Director of the Institute of Biochemical Engineering, School of Chemistry and Chemical Engineering, Beijing Institute of Technology. His research areas include biocatalysis and enzyme engineering, metabolic engineering, and synthetic biology, focusing on the mining, modification, and engineering applications of key enzymes in the synthesis and modification of plant natural products. He has led three projects funded by the National Natural Science Foundation, two sub-projects of the National Key Research and Development Program, and three provincial and ministerial projects. He has published 36 papers as the first author or corresponding author in mainstream journals in the field of biochemical engineering, such as Biotechnology Advances, Natural Product Reports, and Critical Reviews in Biotechnology, and has been granted 11 national invention patents and 1 PCT patent. He was selected for the Beijing Science and Technology New Star Program in 2019. He serves as a young editorial board member of Frontiers of Chemical Science and Engineering and an editorial board member of Synthetic Biology.

For more content, please refer to the bottom 【Read Original】
 About This Journal
About This Journal
The Journal of Biotechnology (founded in 1985, monthly) is jointly sponsored by the Institute of Microbiology, Chinese Academy of Sciences, and the Chinese Society for Microbiology, publicly distributed both domestically and internationally. This journal focuses on reporting the latest research results and progress in the field of biotechnology, with major sections including reviews, industrial biotechnology, synthetic biotechnology, environmental biotechnology, agricultural biotechnology, food biotechnology, and medical biotechnology. The Journal of Biotechnology is a Chinese Core Journal of Peking University, a Core Journal of Chinese Science and Technology, a Core Journal of Chinese Science Citation Database (CSCD), and a journal selected for the Excellent Action Plan for Chinese Science and Technology Journals. The journal has been indexed by several domestic and international databases, including the U.S. Medical Index MEDLINE/PubMed, Netherlands Scopus, U.S. Chemical Abstracts CA, U.S. Biological Abstracts BA, Russian Abstracts AJ, Japan Science and Technology Agency Database JST, U.K. International Agricultural and Biological Sciences Research Abstracts CABI, U.S. Cambridge Scientific Abstracts (Natural Science Volume) CSA(NS), Poland Copernicus Index IC, Netherlands Medical Abstracts EMBASE, Chinese Science Citation Database (CSCD), China National Knowledge Infrastructure (CNKI), Wanfang Database, and more. The Journal of Biotechnology has received numerous honors, including being recognized as one of the Top 100 Chinese Academic Journals (2012), a Chinese Excellent Science and Technology Journal (2014-2017), and receiving the Third Prize in the Science Publishing Fund Technology Journal Ranking by the Chinese Academy of Sciences (2015-2018), as well as support from the Chinese Association for Science and Technology’s Excellent Science and Technology Journal Academic Quality Improvement Project (2015-2017), the Chinese Association for Science and Technology’s Chinese Science and Technology Journal Excellent Construction Plan “Academic Innovation Leading Project” (2018), and the Chinese Science and Technology Journal Excellent Action Plan Project (2019-2023).



Design and Production: Huang Yige
——Produced by the Journal New Media Department