
A public academic platform initiated by overseas scholars
Sharing information, integrating resources
Exchanging academic ideas, occasionally discussing life

In recent years, machine learning algorithms have made significant advancements in many fields, including natural language processing and image recognition. Thanks to the continuous improvement and development of material databases such as Materials Project1, QM92,3, machine learning is increasingly being applied in materials science research. However, due to the specificity of research goals, most work remains limited to solving specific crystal structures and predicting specific material properties. A generalized, universal machine learning model remains a key objective in materials science research. This study is based on the graph neural network framework established by DeepMind.
Based on DeepMind’s established graph neural network framework.

In the field of materials science, the characterization of molecular or crystal structures needs to satisfy translational, rotational, and mirror symmetry, as well as representation of specific information about the overall structure. Common structural features lack universality due to their locality and do not express overall structural information. Graph network models (graph-networks) are structured models based on graph theory that theoretically solve this problem perfectly. In graph theory, a graph is composed of a number of given vertices (nodes) and edges connecting the vertices. When applied to molecular (or crystal) structures, atoms can be described by vertices (nodes), and chemical bonds connecting atoms can be described by edges, thus allowing each molecular or crystal structure to be viewed as an independent “graph”. Based on this type of model, researchers can develop universal models for any material structure or any physicochemical property. Despite the theoretical feasibility, such models are rarely applied in the field of materials science due to model complexity and limitations in material data volume4,5. Recently, the Shyue Ping Ong research group at UC San Diego developed a universal property prediction model for molecules and crystals (MEGNet) based on the graph neural network framework established by DeepMind, achieving leading levels in various property prediction tests6.
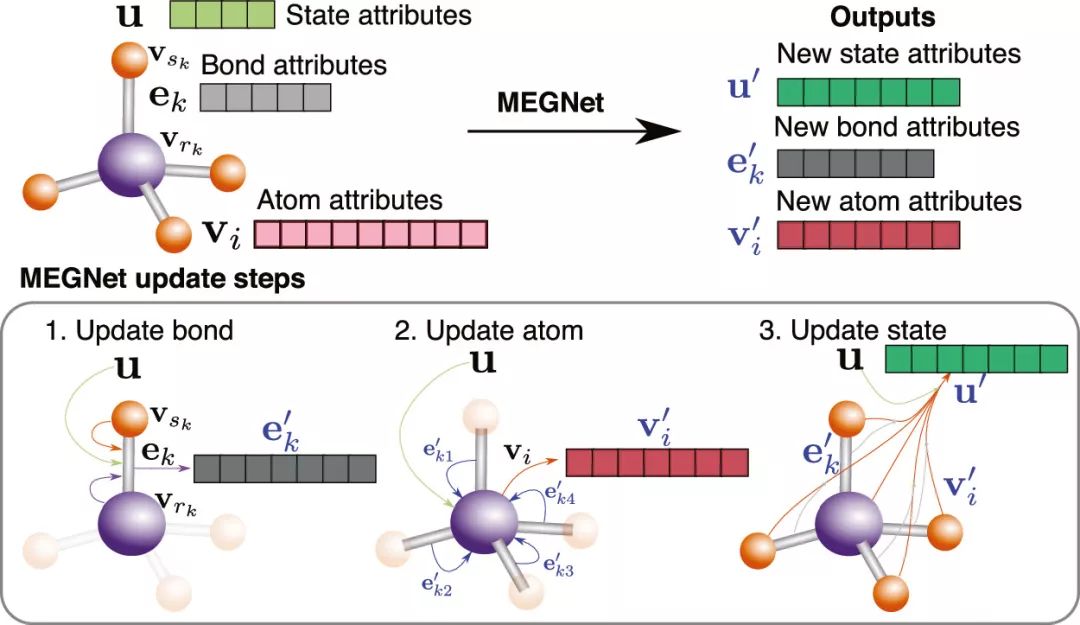
Figure 1. Overview of MEGNet. Each molecular/crystal structure is described by chemical bond information, atomic information, and state information. After each structural description is input into the model, it is updated sequentially until the overall structure’s output properties approach the DFT calculation values.
Figure 1 describes the working mode of the model: each structure can be represented by three vectors, which include atomic information, chemical bond information, and state function information. In each iteration of model training, the chemical bond vector, atomic vector, and state function vector are updated sequentially to obtain a new structural representation vector until the properties output by this representation converge with the DFT calculation results. The authors first trained the model using over 130k data points from the QM9 molecular dataset and predicted 13 physicochemical properties in the molecules, achieving optimal results in 11 of them compared to similar models (Table 1). More advanced is that previous works used separate models for predicting state functions associated with state parameters, such as internal energy (U0, U), enthalpy (H), and Gibbs free energy (G).
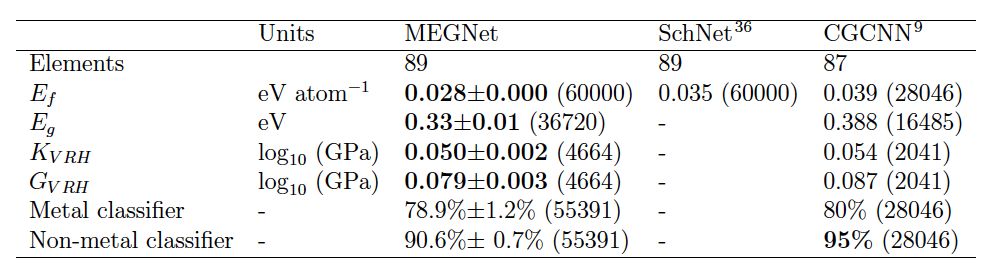
However, in this work, the authors adopted a method of adding state parameters as inputs, allowing a single model to simultaneously predict U0, U, H, and G while maintaining accuracy similar to that of separately trained models, greatly improving training efficiency[YZ1]. In the application to crystal structures, the authors used over 69k data points from the Materials Project database as a training set, performing regression analysis on formation energy, band gap, bulk modulus, and shear modulus, and used band gap values as criteria for classifying metals and non-metals. The mean absolute error (MAE) in the regression analysis was lower than that of similar models SchNet4 and CGCNN5 (Table 2), and the overall accuracy in the classification analysis of metals and non-metals reached 86.9%, with an AUC of 0.926 in the ROC curve, comparable to the previously optimal model CGCNN.
Table 1. Comparison of Mean Absolute Errors (MAE) of Different Models in Predicting 13 Properties on QM9
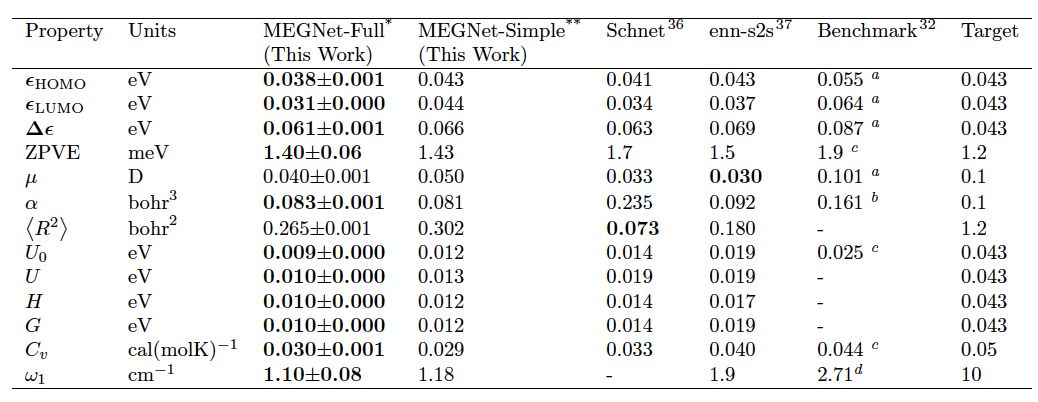
Table 2. MEGNet and Other Graph-Based Models
Comparison of Prediction Accuracy on the Materials Project Dataset
In an in-depth analysis of the model, the authors found that the element embeddings extracted from the optimal model corresponded with chemical knowledge. For example, projecting the element embeddings into two-dimensional space revealed that Eu and Yb are farther from other lanthanide elements and closer to alkaline earth metals, which aligns with chemical experience. This kind of analysis not only supports that the model can learn reliable chemical information but also allows the learned chemical information to be used for transfer learning, significantly reducing the amount of data needed to train new models. For instance, in this case, the authors used the element embeddings extracted from a model trained with ~69k formation energy data to predict the band gap and elastic properties, where the data volume was only half or even one-tenth of that for formation energy. By using transfer learning, the authors achieved lower MAE and doubled the convergence speed compared to direct training. This provides a feasible solution for efficient and accurate model training with small data volumes.
In terms of model usage, users can log in to http://megnet.crystals.ai, enter the crystal structure code or cif file according to the prompts to obtain the predicted properties from the model. Moreover, all the Python code involved in the article has been open-sourced (https://github.com/materialsvirtuallab/megnet.git). Below are examples of how to use the existing model and train a new model.
1Example 1: Using the Molecular Model

2Example 2: Using the Crystal Model to Predict Shear Modulus

3Example 3: Training a New Model

(1) Jain, A.; Ong, S. P.; Hautier, G.; Chen, W.; Richards, W. D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A Materials Genome Approach to Accelerating Materials Innovation. APL Mater. 2013, 1 (1), 011002. https://doi.org/10.1063/1.4812323.
(2) Ruddigkeit, L.; van Deursen, R.; Blum, L. C.; Reymond, J.-L. Enumeration of 166 Billion Organic Small Molecules in the Chemical Universe Database GDB-17. J. Chem. Inf. Model. 2012, 52 (11), 2864–2875. https://doi.org/10.1021/ci300415d.
(3) Ramakrishnan, R.; Dral, P. O.; Rupp, M.; von Lilienfeld, O. A. Quantum Chemistry Structures and Properties of 134 Kilo Molecules. Sci. Data 2014, 1, 140022. https://doi.org/10.1038/sdata.2014.22.
(4) Schütt, K. T.; Sauceda, H. E.; Kindermans, P.-J.; Tkatchenko, A.; Müller, K.-R. SchNet – A Deep Learning Architecture for Molecules and Materials. J. Chem. Phys. 2018, 148 (24), 241722. https://doi.org/10.1063/1.5019779.
(5) Xie, T.; Grossman, J. C. Crystal Graph Convolutional Neural Networks for an Accurate and Interpretable Prediction of Material Properties. Phys. Rev. Lett. 2018, 120 (14), 145301. https://doi.org/10.1103/PhysRevLett.120.145301.
(6) Battaglia, P. W.; Hamrick, J. B.; Bapst, V.; Sanchez-Gonzalez, A.; Zambaldi, V.; Malinowski, M.; Tacchetti, A.; Raposo, D.; Santoro, A.; Faulkner, R.; et al. Relational Inductive Biases, Deep Learning, and Graph Networks. ArXiv180601261 Cs Stat 2018.
(7) Chen, C.; Ye, W.; Zuo, Y.; Zheng, C.; Ong, S. P. Graph Networks as a Universal Machine Learning Framework for Molecules and Crystals. Chem. Mater. 2019. https://doi.org/10.1021/acs.chemmater.9b01294.
Further Reading
npj: Machine Learning—Rapid and Accurate Prediction of Electronic Structure Problems
npj: Deep Learning Prediction—Band Gap of Hybrid Graphene-Boron Nitride Structures
npj: High Entropy Alloys—Yield Strength Prediction Based on First Principles
npj: Machine Learning—Neural Network Methods for Calculating Formation Energies of Multicomponent Crystals
This article is part of the special content of NetEase News – NetEase Account “Each Has Its Attitude”
For media reprint authorization, please see below
