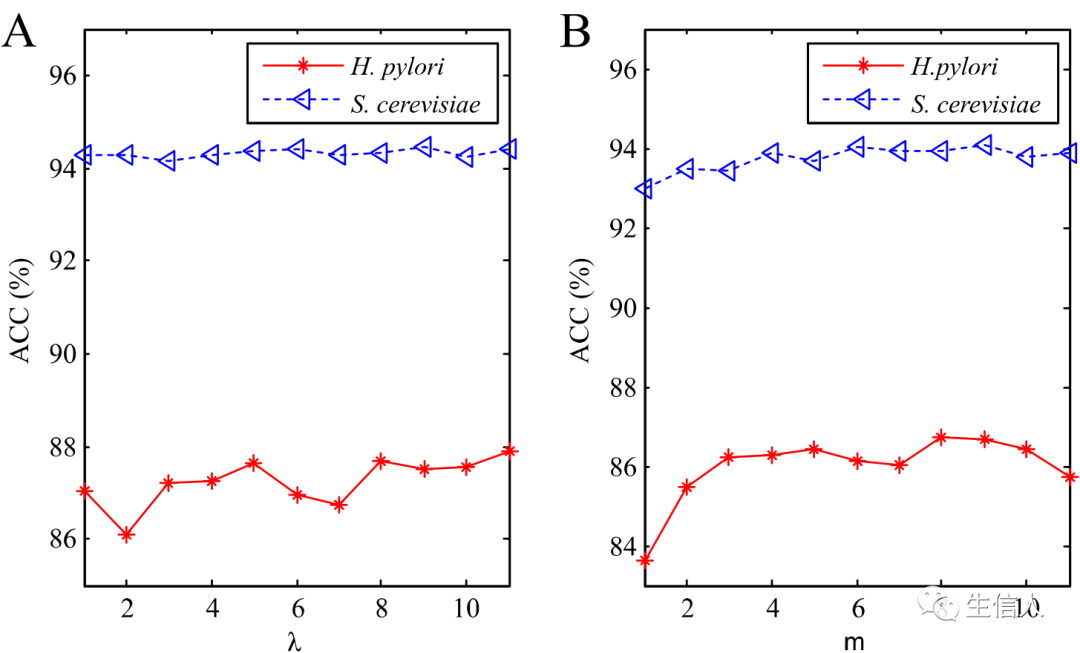
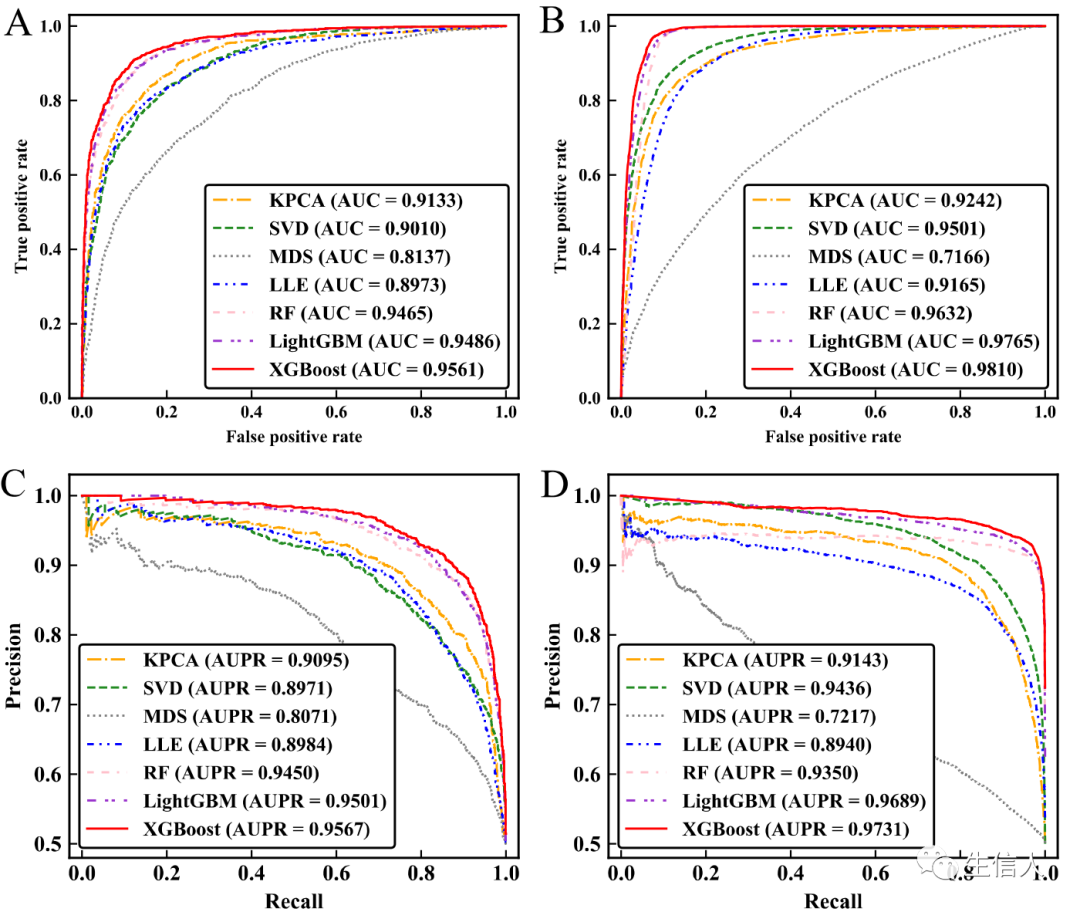
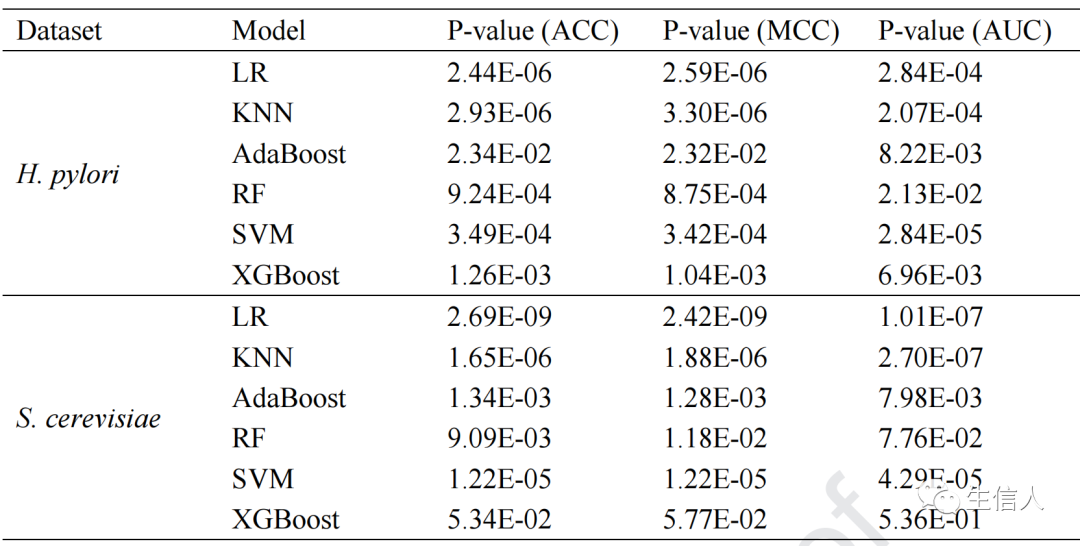
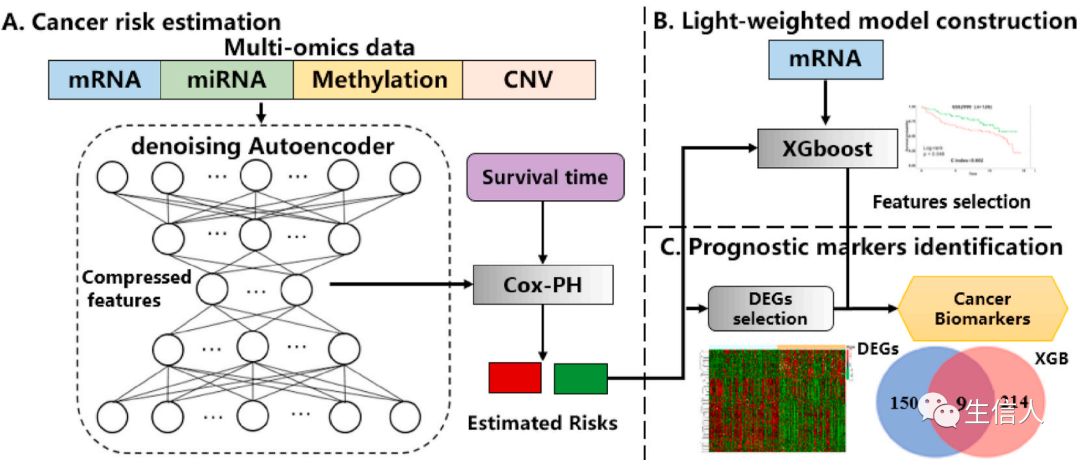
 , then the strong learner of Boosting can be represented as:
, then the strong learner of Boosting can be represented as:







END
Previous Recommendations
|
Character Specials |
• Comprehensive summary of academician Dong Chen’s research results • Series of research results by Professor Zhang Zemin |
|
Immunity Specials |
• Pure bioinformatics mining of immunotherapy in Division 2 • Multi-omics immunotherapy |
|
Spatial Metabolism |
• Single-cell sequencing combined with spatial transcriptomics • Spatial metabolism + drug resistance |
|
Iron Death Specials |
• Summary of iron death articles • Classic iron death, new ideas again |
|
m6A Specials |
• Integration of m6A regulatory factors in bioinformatics analysis • Why does m6A glioma continue to rise? |
|
Research Acceleration |
• How bioinformatics accelerates research topics • Essential skills for clinical research wet experiments |
|
irAEs Specials |
• irAEs: Overview • irAEs: Bioinformatics signatures |