
White Dot Syndromes (WDS) are a group of inflammatory diseases affecting the outer layers of the retina, retinal pigment epithelium (RPE), and/or choroid. WDS includes: Birdshot Chorioretinopathy (BCR), Multifocal Erased White Dot Syndrome (MEWDS), Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE), Multifocal Choroiditis with Pan-Uveitis (MCP), Serpiginous Choroiditis (SC), Punctate Inner Choroidopathy/ Multifocal Choroiditis (PIC/MFC), and Persistent Placoid Chorioretinitis (RPC).
Seeing these names, are you as confused as I am? Fortunately, the development of multimodal imaging technology allows us to better understand the lesion morphology and activity of the disease, and classify them anatomically. Professor Meisha L. Raven from the Department of Ophthalmology and Visual Sciences at the University of Wisconsin published a review on White Dot Syndromes in the International Journal of Retina and Vitreous that surely clears up our confusion. Let’s get started!

White Dot Syndromes (WDS) are a group of posterior uveitis diseases with similar clinical features, though they are unique diseases with some overlap. Other types of uveitis, tumors, and inflammation can also “disguise” themselves as White Dot Syndromes when they involve the choroid. The rapid development of multimodal imaging technology in recent years has made precise anatomical localization of lesions possible. Combining SD-OCT, EDI mode SD-OCT imaging, FFA, ICGA, ultrasound imaging, wide-angle imaging, and autofluorescence (AF) along with the latest OCTA for lesion localization may be more appropriate for anatomically classifying these diseases rather than simply categorizing them as “White Dot Syndromes.” These types include:
-
Uveitis Syndromes with Choroidal Pathological Changes
Sympathetic Ophthalmia
-
Systemic Uveitis Syndromes with Choroidal Pathological Changes
Vogt–Koyanagi–Harada Disease
Sarcoidosis
-
Multifocal Choroiditis with Outer Retinal/Choroidal Capillary Changes
Without Vitritis
Histoplasmosis
Punctate Inner Choroidopathy (PIC)
With Vitritis
Multifocal Choroiditis with Pan-Uveitis (MCP)
Multifocal Erased White Dot Syndrome (MEWDS)
Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE)
Serpiginous Choroiditis (SC)
Persistent Placoid Chorioretinitis (RPC)
Birdshot Chorioretinopathy (BCR)
-
Tumors
Primary Choroidal Lymphoma
Primary Vitreoretinal Lymphoma
Secondary (Metastatic) Lymphoma
-
Infections: Syphilis, Tuberculosis, Lyme Disease


1、Sympathetic Ophthalmia
Sympathetic Ophthalmia (SO) is a bilateral granulomatous uveitis that occurs weeks to decades after penetrating ocular injury or surgical trauma.
Anterior Segment: Anterior chamber cells and posterior keratic precipitates (KP), iris thickening, and posterior synechiae;
Posterior Segment: Vitritis, exudative retinal detachment, and disc edema. The classic fundus appearance is Dalen-Fuchs nodules, which appear as yellow-white lesions with focal elevation between the retinal RPE and Bruch’s membrane. Late stages may show “sunset glow” fundus secondary to choroidal pigment loss;
Ultrasound: Choroidal thickening, possibly with exudative retinal detachment.
SD-OCT: Diffuse choroidal thickening, subretinal fluid, irregular inner/outer segment (IS/OS) junction, and external membrane. FFA: Disc leakage and multiple hyperfluorescent leakage points at the RPE level. Early focal blockage may occasionally be seen.
ICGA: Multiple hypofluorescent areas with dye accumulation in late stages.
Treatment: Prognosis significantly improves with the use of corticosteroids and immunosuppressants. Early treatment of the disease is essential to prevent significant vision loss.
Key Differentiation Points: Ocular trauma, multiple surgical history, bilateral anterior and posterior segment inflammation, Dalen-Fuchs nodules on fundus.

2、 Vogt–Koyanagi–Harada Disease
VKH is a bilateral granulomatous uveitis. It is usually associated with exudative retinal detachment and extraocular manifestations such as pleocytosis in cerebrospinal fluid, tinnitus, hearing loss, and skin changes (such as hair loss and vitiligo). This disease is more common in people of color, such as Asians, Latinos, Native Americans, and Asian Indians.
Clinical Manifestations: Early patients often experience prodromal symptoms including fever, nausea, headache, dizziness, orbital pain, photophobia, and meningitis. This is followed by bilateral blurred vision secondary to posterior uveitis.
Anterior Segment: Inflammation extends to the anterior segment, with anterior chamber cells and KP.
Posterior Segment: Vitritis, retinal choroidal edema, multifocal retinal pigment epithelium detachment, and late stages show “sunset glow” fundus with choroidal retinal atrophy in the peripheral fundus.
B-scan: Shallow anterior chamber, choroidal shallow detachment, ciliary body thickening, and serous retinal detachment.
EDI-OCT: Choroidal thickening, serous retinal detachment.
FA: Early multiple pinpoint leakage points, followed by multiple focal leaks, and late stages show subretinal dye accumulation.
FAF: Peripheral retinal abnormalities may be seen in chronic VKH patients.
ICGA: Early choroidal vascular hyperfluorescence and vascular leakage, with low fluorescence appearing at the choroidal level in late stages. High fluorescence staining may be seen at the disc.
Treatment: Long-term use of corticosteroids, usually for more than 6 months. Uncontrolled cases should be supplemented with immunomodulatory therapy. Early treatment yields better visual prognosis for patients.
Key Differentiation Points:Prodromal symptoms, multifocal neuroepithelial detachment, FA pinpoint leakage, and lake-like fluorescence accumulation.
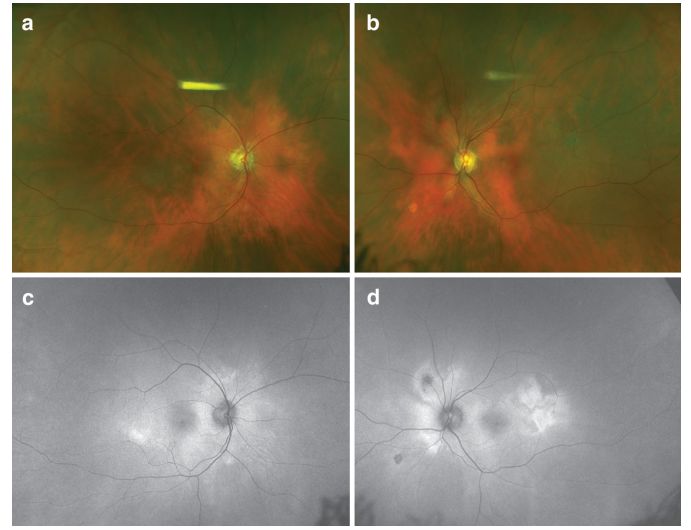
Image: “Sunset glow” fundus in VKH patients (a-b). AF shows patchy high autofluorescence (c-d).

3、 Sarcoidosis
Sarcoidosis is an idiopathic granulomatous disease that involves multiple systems, with 15-30% of patients having ocular involvement. It is most common in African Americans. It can affect the orbit, ocular appendages, anterior and/or posterior segments.
Anterior Segment: Anterior uveitis, iris nodules, conjunctival nodules, and scleral nodules.
Posterior Segment: 60% of patients with ocular involvement show posterior segment lesions, including vitritis, choroidal retinitis, vascular occlusion, vascular sheathing, neovascularization, and disc granulomas. Vitreous inflammation may aggregate to form “pearl necklace” or “snowball” changes. Choroidal granulomas are commonly seen in peripheral or mid-peripheral retina, with severe choroidal granulomas presenting as “wax candle drop” lesions, and choroidal granulomas manifesting as isolated or multifocal yellowish round elevated lesions beneath the retina. Large granulomas may be associated with serous retinal detachment.
EDI-OCT: Choroidal low reflective thickening.
FFA: Early lesions show hypofluorescence, isofluorescence, and late-stage fluorescence staining shows hyperfluorescence.
ICGA: Choroidal granulomas show low fluorescence.
Treatment: Use topical corticosteroid eye drops to treat anterior chamber inflammation. Mydriatic eye drops relieve ciliary muscle spasm and prevent posterior synechiae formation. Posterior segment inflammation is usually treated with sub-Tenon’s capsule corticosteroid injection, intravitreal triamcinolone (TA), corticosteroid implants, or immunotherapy.
Key Differentiation Points:Systemic granulomas, bilateral involvement, iris nodules, “pearl necklace” or “snowball” opacities, choroidal nodules.

4、 Ocular Histoplasmosis Syndrome
The ocular histoplasmosis syndrome (OHS) is a chorioretinal disease caused by histoplasmosis infection.
Clinical Manifestations: After initial exposure to the fungus, patients may experience mild flu-like symptoms and asymptomatic calcified pulmonary nodules. Years after systemic infection subsides, when CNV occurs, patients typically experience decreased vision, metamorphopsia, and paracentral scotomas.
Anterior Segment: No inflammation;
Posterior Segment: No posterior segment inflammation, characteristic “punched-out” chorioretinal scars. The classic triad includes peripheral and posterior ocular histoplasmosis spots (“histo spots”), peripapillary chorioretinal atrophy (PPA), and CNV. At least two of the three features must be present to diagnose OHS.
OCT: Affected outer retinal areas show high reflective chaos as “histo spots”.
FFA: In asymptomatic patients, a high reflective early window defect pattern is observed, with late-stage peripheral atrophy spots and progressive staining of macular scars. If subretinal fluid or subretinal hemorrhage occurs, early high fluorescence and late leakage of small vessels in the subretinal or RPE space can diagnose CNV.
ICGA: Occult CNV shows early high staining.
FAF: OHS lesions show round low autofluorescence.
Diagnostic Points:History of histoplasmosis, triad (peripapillary scarring, “histo spots”, CNV in macular area).

5、 Punctate Inner Choroidopathy (PIC)
PIC is characterized by multiple deep yellow-white punctate nodular lesions in the posterior pole and progressive atrophy, an inflammatory disease of the choroid and outer retina.
Clinical Manifestations: Decreased vision, paracentral scotomas, and metamorphopsia.
Anterior Segment: No anterior uveitis;
Posterior Segment: No signs of vitritis. In the acute phase, multiple small (100-300μm), well-defined yellow-white lesions are seen in the posterior pole. The lesions may merge to form serous retinal detachment. They may also gradually atrophy to form yellow-white chorioretinal scars, with mild disc edema. 40-76% of patients may show CNV at initial diagnosis.
OCT: RPE focal elevation, breaking through the external membrane and outer nuclear layer, forming “hump” like moderate reflective nodular lesions. If CNV exists, OCT may show intraretinal fluid. OCTA can detect CNV. Focal thinning of the choroid adjacent to PIC lesions.
FFA: The lesions show more than what is found in clinical examination, with early hyperfluorescence and late staining. Atrophic lesions appear as window defects.
FAF: Active lesions show weak spontaneous fluorescence with a ring of high fluorescence edge.
ICGA: Lesions show low fluorescence, with the same quantity as FFA.
ERG and EOG show no significant changes.
Prognosis: CNV and CME can lead to significant vision impairment. Active treatment of CNV can maintain vision of 20/40 or better. Recurrence rates are 33-66.7%.
Key Differentiation Points:Multiple yellow-white creamy lesions in the posterior pole of the fundus, no anterior or posterior segment inflammation, “hump” like moderate reflective nodular lesions on OCT in the outer retina.
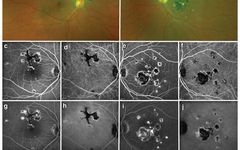
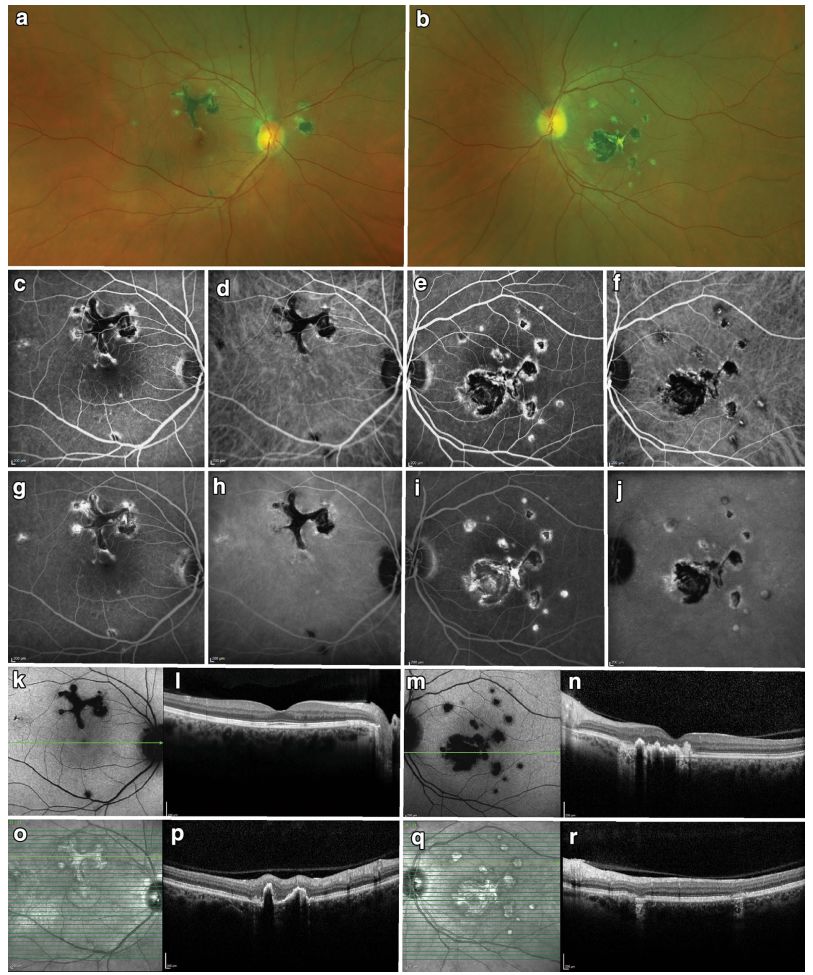 Image: PIC, bilateral multifocal lesions (a-b). FA shows early (c, e) low fluorescence lesions with slight high reflective edges, gradually staining over time (g, i). ICGA shows similar morphology but generally low fluorescence (d, h, f, j). Spontaneous fluorescence of the fundus shows (k, m) low spontaneous fluorescence of the lesions. Infrared imaging shows high reflectivity of lesions (o, q). OCT scans of the fovea are normal (l), but B-scans above the fovea show localized elevations of the outer retina and PRE (p). The lesions in the left eye are similar (n, r).
Image: PIC, bilateral multifocal lesions (a-b). FA shows early (c, e) low fluorescence lesions with slight high reflective edges, gradually staining over time (g, i). ICGA shows similar morphology but generally low fluorescence (d, h, f, j). Spontaneous fluorescence of the fundus shows (k, m) low spontaneous fluorescence of the lesions. Infrared imaging shows high reflectivity of lesions (o, q). OCT scans of the fovea are normal (l), but B-scans above the fovea show localized elevations of the outer retina and PRE (p). The lesions in the left eye are similar (n, r).

dvdf
6、 Multifocal Choroiditis with Pan-Uveitis (MCP)
MCP is a chronic, bilateral disease, usually affecting young healthy individuals, especially 30-50 year old myopic women.
Clinical Manifestations: Decreased vision, dark spots, and photopsia. Patients often experience periodic flare-ups, which can distinguish it from ocular histoplasmosis.
Anterior Segment: Anterior chamber cells and KP;
Posterior Segment: During the acute phase, multiple round, poorly defined yellow-white lesions can be seen in the posterior pole and periphery. The lesions eventually evolve into “punched-out” scars with pigmented borders. During active phases, disc congestion, retinal vasculitis, and CME may be observed.
OCT: RPE-like glass membrane substance beneath the lesions, with high reflectivity in the choroid beneath the lesions.
FFA: Lesions during the acute phase show early fluorescence masking, with high fluorescence staining in late stages. CME and CNV may also be displayed.
ICGA: Shows low fluorescence changes in choroidal lesions.
FAF: Low spontaneous fluorescence changes in the posterior pole and periphery.
OCTA can detect CNV.
Multifocal ERG: Shows significant functional abnormalities in the measurement areas.
Prognosis: This disease has a high recurrence rate. Vision prognosis is poor due to macular scarring, atrophy, or chronic CME. Topical or periorbital corticosteroids are used for treatment, and systemic immunosuppressants are applied when necessary. Secondary CNV requires anti-VEGF medication treatment.
Key Differentiation Points: Symmetrical bilateral multifocal choroidal lesions, anterior and posterior segment inflammation, OCT shows RPE-like glass membrane substance beneath the lesions, high recurrence rate.

7、 Multifocal Erased White Dot Syndrome (MEWDS)
MEWDS is an acute multifocal disease primarily affecting one eye, prevalent in myopic young individuals, with a female-to-male ratio of 5:1.
Clinical Manifestations: Many have flu-like prodromal symptoms. Blurred vision, photopsia, color blindness, temporal visual field defects, central or temporal scotomas. Visual field tests may show an enlarged physiological blind spot.
Anterior Segment: No anterior chamber inflammation;
Posterior Segment: Mild vitritis may be observed. The typical fundus feature is poorly defined yellow-white lesions predominantly in the macular region, extending into the mid-peripheral retina. The lesions usually resolve within weeks to months, with rare recurrence.
OCT: Shows destruction of the ellipsoid zone, with high reflective material accumulating beneath the RPE and in the ellipsoid and outer nuclear layers.
FFA: Early lesions appear as round or ring-shaped strong fluorescent spots;
ICGA: Shows multiple small round low fluorescence points, with more spots than those seen on FFA.
FAF: High spontaneous fluorescence in the acute phase. Wide-angle FAF shows acute phase lesions appearing in the posterior pole and spreading to the periphery; then the lesions gradually disappear centripetally from the periphery.
ERG shows reduced a-wave amplitude. EOG may also be abnormal. After disease remission, ERG and EOG tend to normalize.
Prognosis: MEWDS has a good prognosis. Some patients may experience persistent enlargement of blind spots, flashes, and color vision disturbances. Most cases self-resolve and generally do not require treatment, with rare recurrence.
Key Differentiation Points: Acute onset, unilateral involvement, poorly defined white dot lesions in the posterior pole and mid-periphery, enlarged physiological blind spot, self-limiting, strong fluorescent spots on FFA, destruction of the ellipsoid zone on OCT.
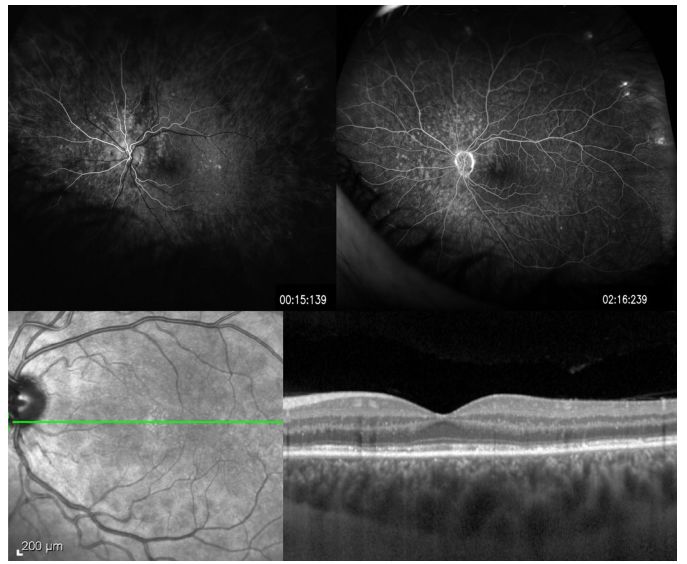
Image: MEWDS, a 26-year-old female patient with mild vision decrease and photopsia. FFA shows early high fluorescence lesions in a ring structure (upper left), continuing to the late phase (upper right). OCT shows ellipsoid destruction and high reflective material accumulation above the RPE, extending through the crossing zone, ellipsoid, and outer nuclear layers (lower right).

8、 Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE)
APMPPE is an acutely occurring inflammatory disease that can affect the choroidal capillaries, RPE, and outer retina. 70% of patients involve the opposite eye within days or weeks after initial onset. The cause is still unclear.
Clinical Manifestations: Sudden bilateral painless vision decrease.
Anterior Segment: No anterior segment inflammation;
Posterior Segment: Mild to moderate vitritis. The posterior pole may show numerous gray-white lesions, placoid, with irregular edges, poorly defined, rarely seen equatorially.
OCT: In early stages, shows high reflective outer retina, with the high reflectivity of the outer retina decreasing as the lesions resolve. Destruction of the IS/OS junction and RPE atrophy may persist.
FFA: Early lesions show low fluorescence, with strong fluorescent spots with poorly defined edges in late stages.
ICGA: More low fluorescence lesions than those observed with the ophthalmoscope.
FAF: Lesions show low spontaneous fluorescence, appearing later, and fewer than what is clinically observed in APMPPE.
OCTA: Shows areas of low choroidal perfusion corresponding to changes seen in ICGA.
ERG shows mild abnormalities in a-wave and b-wave amplitudes during the acute phase. EOG is also abnormal during the acute phase, but improves within 2-3 months after disease remission.
Prognosis: This is a self-limiting disease, usually with a good visual prognosis. Involvement of the fovea, older age, unilateral involvement, and recurrence may lead to poor visual prognosis.
Key Differentiation Points: Bilateral posterior pole multifocal gray-white lesions, placoid, irregular edges, poorly defined, no anterior segment and posterior segment inflammation, early low fluorescence on FFA, poorly defined strong fluorescence in late stages.

9、 Serpiginous Choroiditis (SC)
SC is a chronic and progressive disease affecting both eyes, which is relatively rare, with a slightly higher incidence in males.
Clinical Manifestations: Unilateral central vision decrease, distortion, or scotomas. Often recurrent.
Anterior Segment: Inflammation is usually mild;
Posterior Segment: The vitreous may be clear or show mild inflammation. In the acute phase, gray-white or yellow lesions around the disc may be observed, slightly elevated, involving choroidal capillaries and RPE. New active lesions are located at the edges of old lesions, expanding irregularly outward from around the disc in a serpentine or spiral manner.
OCT: Active lesions show high reflectivity and thickening of the outer retina. Both active and inactive lesions will show destruction of the IS/OS junction.
FFA: Active lesions show early low fluorescence and late high fluorescence. Old lesions show window defects and late staining.
ICGA: Active lesions show map-like merging or patchy low fluorescence in early and mid-stages, with low fluorescence or fluorescence consistent with the background in late stages.
FAF: New high spontaneous fluorescence appears at the edges of low fluorescence old lesions.
Electrophysiological examinations are usually normal.
Key Differentiation Points: Bilateral involvement, anterior segment or vitreous inflammation, choroidal lesions expanding from around the optic disc in a serpentine or spiral manner, with new yellow-white lesions appearing at the edges of old lesions.
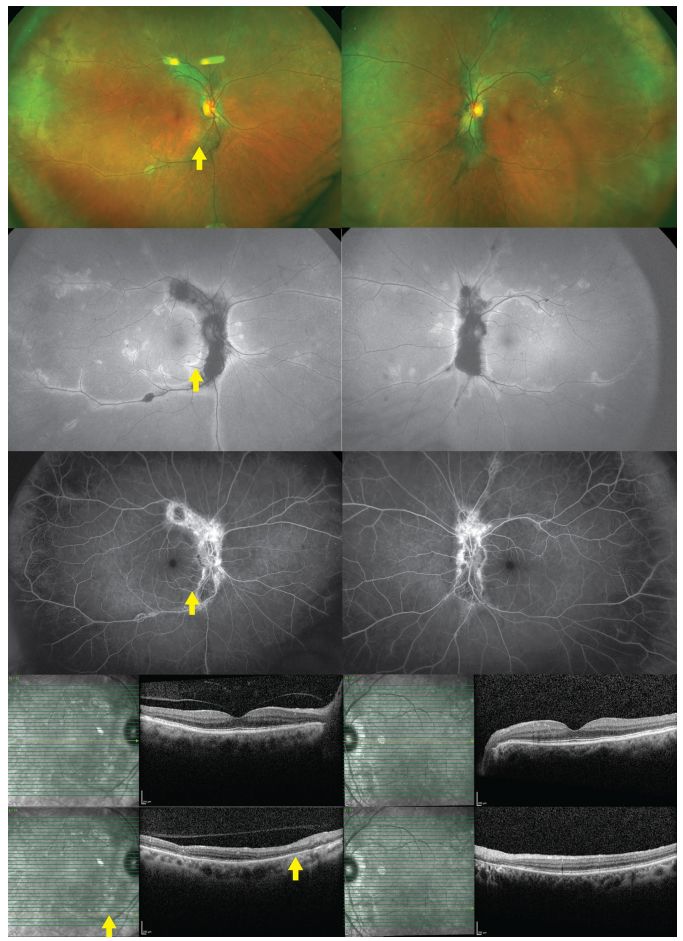
Image: Serpiginous Choroiditis. A female patient with chronic visual field loss in both eyes. FAF shows low spontaneous fluorescence centrally with high spontaneous fluorescence at the edges (second row). FFA shows high fluorescence staining with central patchy low fluorescence (third row), OCT shows a normal fovea, but the nasal elliptical area shows destruction of the outer membrane and retinal pigment epithelium.

10、 Persistent Placoid Chorioretinitis (RPC)
RPC is a chronic recurrent disease of unknown etiology.
Clinical Manifestations: Sudden painless vision decrease, distortion, and central scotomas without apparent cause. This is often confused with APMPPE and SC. A duration of more than 6 months is often key to its diagnosis and differentiation from APMPPE.
Anterior Segment: Uveitis;
Posterior Segment: Mild vitreous inflammation, bilateral posterior pole creamy white lesions, approximately half the diameter of the optic disc, with over 50 to several hundred lesions in different stages of the disease, which is characteristic. The difference between RPC and MEWDS is that RPC tends to involve the peripheral retina.
OCT: High reflectivity of the inner and outer retina during the active phase.
FFA: Early lesions show low fluorescence and late staining.
FAF: Extensive low spontaneous fluorescence in the posterior pole and mid-periphery,
ICGA: Lesions show low fluorescence in early and late stages.
ERG and EOG are normal.
Key Differentiation Points: Bilateral milky white acute lesions and old pigment changes, with a total number of lesions exceeding 50 to several hundred. Lesions may occur in the peripheral retina, and new lesions do not necessarily appear at the edges of old lesions.
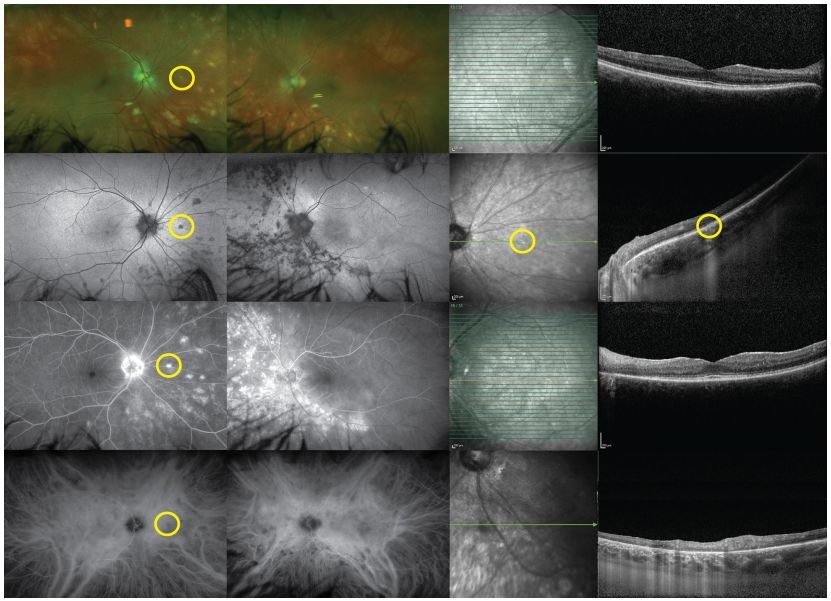
Image: RPC. Fundus images (left) show distinct stages of chorioretinal scarring. FFA (right) shows late-stage chorioretinal scarring staining, along with low fluorescence areas of pigment deposition.
11、 Birdshot Chorioretinopathy (BCR)
BCR is a bilateral chronic uveitis characterized by multiple creamy lesions of the choroid. The cause of BCR is still unclear.
Clinical Manifestations: There is a slight female predominance, usually seen in patients aged 40 to 60, with nearly 90% of patients being HLA-A29 antigen positive. Symptoms include blurred vision, vision loss, photophobia, and photopsia.
Anterior Segment: Anterior segment inflammation is usually absent.
Posterior Segment: Mild to moderate vitreous inflammation, multiple oval or round creamy lesions in the choroid, usually concentrated around the optic disc, radiating towards the periphery, most commonly seen below and nasally to the optic disc. Retinal vasculitis is characterized by retinal vascular narrowing. CME and disc edema may also be present.
EDI-OCT: Disruption of the photoreceptor IS/OS junction.
FFA: Early lesions show low fluorescence, while late stages show high fluorescence. High fluorescence at the disc, vascular leakage, and late CME with prolonged arteriovenous transit time are unique to BCR.
FAF: Chorioretinal atrophy areas show low spontaneous fluorescence.
ICGA: More lesions than observed under the ophthalmoscope show weak fluorescence, with strong fluorescence spots appearing in late stages.
ERG: 88.8% of patients show abnormalities, making it an important tool for monitoring disease progression; EOG is generally normal.
Visual: Visual field narrowing, enlarged blind spot, central or paracentral scotomas.
Prognosis: Due to the chronic nature of the disease, it often relapses multiple times, leading to poor visual prognosis in late stages. Loss of photoreceptors, macular edema, and disc edema leading to disc atrophy are the main causes of vision loss. Corticosteroids can be used in the short term.
Key Differentiation Points: Bilateral involvement, HLA-A29 antigen positive, retinal vasculitis, macular cystoid edema, and at least three choroidal lesions around the optic disc.
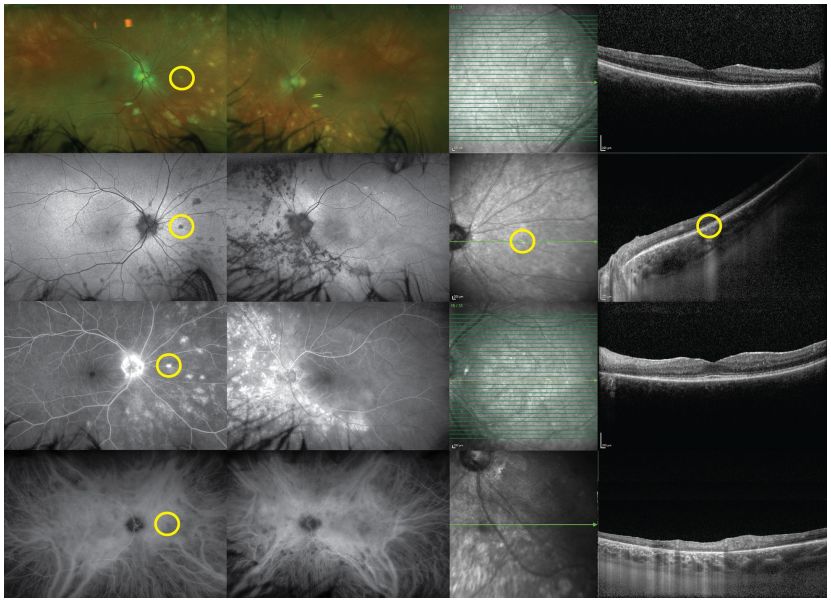
Image: BCR, fundus examination reveals deep, multifocal, white choroidal lesions, primarily located nasally (upper row). FAF shows low spontaneous fluorescence of the lesions (second row). Lesions appear as high fluorescence on FFA (third row), corresponding to low fluorescence points on ICGA (fourth row).
12、 Tumors
Intraocular lymphoma includes primary vitreoretinal lymphoma (PVRL), primary choroidal lymphoma, and secondary (metastatic) lymphoma. Primary vitreoretinal lymphoma (PVRL) affects the vitreous and RPE. The choroidal tissue is the primary site of metastatic and choroidal lymphoma. Most patients present with painless vision loss;
PVRL is the most common intraocular lymphoma and is often associated with central nervous system diseases. It is common in elderly or immunocompromised patients. Most cases are bilateral but may present asymmetrically.
Anterior Segment: Inflammation is rare.
Posterior Segment: The presence of aggregated vitreous cells and multiple irregular yellow-white deposits beneath the RPE is a hallmark of the disease. Disc scarring, retinal vasculitis, solid retinal pigment epithelial detachment, or exudative retinal detachment may be present.
OCT: Shows exudation beneath the RPE and PED.
EDI-OCT is particularly suitable for choroidal lymphoma with thin-layer tumor infiltration, which may show a “calm, undulating, or stormy (choppy)” appearance depending on the thickness of the invasion.
FFA: Retinal deposits show fluorescence staining, RPE window defects, and diffuse RPE granularity.
FAF: RPE deposits show high spontaneous fluorescence, while subretinal deposits show low fluorescence.
Primary choroidal lymphoma and secondary (metastatic) lymphoma usually present unilaterally.
Anterior Segment: Anterior chamber cells and KP are rare.
Posterior Segment: Most patients have vitreous cells. Fundus examination reveals multiple yellow retinal sublesions that may lead to RPE separation. Choroidal creamy thickening and RPE proliferation may also be present.
OCT: Shows tumor cell aggregation beneath the RPE;
FFA: Round low fluorescence lesions;
Ultrasound: Choroidal thickening, subretinal masses, and vitreous opacities.
ICGA: Shows low fluorescence areas corresponding to clinically observed choroidal inflammation.

13、 Infections
Syphilitic fundus lesions manifest as yellow, patchy chorioretinal lesions located in the posterior pole or macula.
OCT: Shows IS/OS junction and external membrane defects.
FFA: Early low fluorescence areas may be seen, with late staining resembling a persistent low fluorescence background like a leopard’s spots. ERG may be significantly reduced.
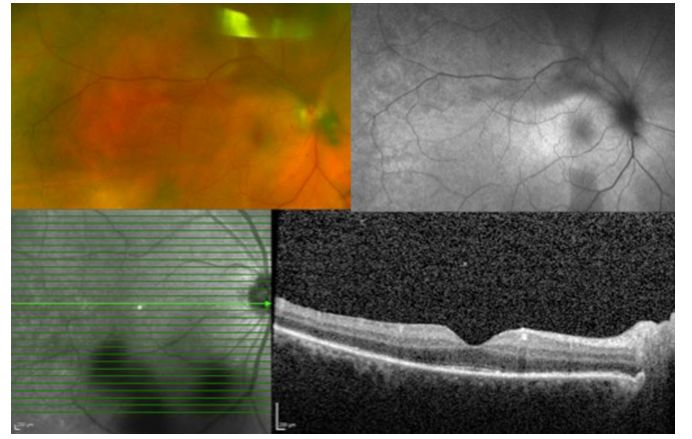
Image: Syphilitic uveitis, pan-uveitis in the right eye; OCT (below) shows segmental loss in the ellipsoid zone.
Ocular tuberculosis (TB) may show choroidal nodules, multifocal choroiditis, and serpiginous choroiditis. Miliary choroidal nodules present as yellow-white small tumors with poorly defined boundaries at the choroidal level, which may develop into a single tuberculoma. OCT: There are local adhesions between the PRE-choroidal capillary layer and the neural sensory retina.
FFA: Choroidal nodules show low fluorescence in early stages and high fluorescence in late stages. Healed nodules present with translucent high fluorescence reflections. Choroidal tuberculomas show early high reflectivity, with rapid fluorescence enhancement in the late stage due to exudative retinal detachment.
ICGA: Acute choroidal lesions show low fluorescence in early and mid-stages, with multiple small strong fluorescence areas appearing in late stages.
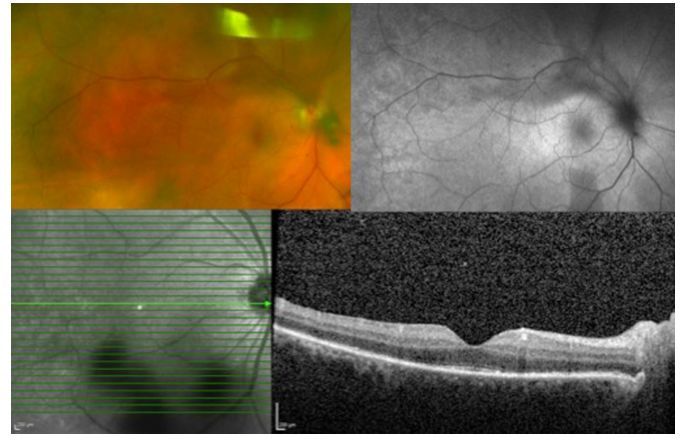
Image: Pulmonary tuberculosis secondary to pan-uveitis. Color fundus photograph (upper left) shows patchy choroidal atrophy. FFA (right upper), ICGA (left lower), and FAF (right lower) show low fluorescence areas corresponding to areas of chorioretinal atrophy.
Patients with ocular Lyme disease are characterized by photophobia and severe periodic ocular pain. The choroiditis caused by this disease presents as multiple localized creamy lesions seen deep in the retina, accompanied by pigmentation and choroidal atrophy. FFA shows low fluorescence of the lesions, with granular high fluorescence surrounding the lesions.

Given the similar histories, clinical presentations, and imaging characteristics among diseases, diagnosing White Dot Syndromes poses significant challenges for many clinicians. Accurate diagnosis is crucial for guiding disease treatment, visual prognosis, and disease monitoring. Multimodal imaging technologies such as SD-OCT, EDI-OCT, FFA, ICGA, ultrasound, wide-angle imaging, AF, and OCTA can clarify anatomical localization of lesions, helping doctors differentiate diseases and establish clinical diagnoses. We hope this review will be beneficial to clinical work.
Original link:
https://www.ncbi.nlm.nih.gov/pubmed/?term=Multi%E2%80%91modal+imaging+and+anatomic+classification+of+the+white+dot+syndromes