Author | Cheng Yu
Reviewer | Zhu Yulei

Today, I would like to introduce a paper jointly published in January 2020 by Bayer Crop Science, Bayer’s Machine Learning R&D Department, and the Genetic Toxicology Department in Nature Communications. This paper discusses a generative model for de novo design and synthetic optimization of molecules. The generative model is trained using transcriptomic data, which allows for the automatic generation of molecules with the desired transcriptomic profiles with high probability. By providing only the gene expression markers of the desired state without any prior target annotation for trained compounds, the model can automatically design molecules with similar activity for the desired targets, resulting in molecules that are more similar to active compounds, offering a new approach for drug development.
1. Background
Drug development is a long and arduous process, with the challenge being that only a small fraction of the theoretically possible 1060 drug-like molecules can be used for treatment. One of the challenges faced in the drug discovery process is the identification of hit compounds. Hit compounds refer to molecules that exhibit preliminary activity against specific targets or mechanisms, marking the starting point of the entire drug discovery process. Hit compounds may be natural ligands or molecules selected from literature, patents, or structural information of molecules. However, for novel or isolated targets with no prior information available, previous methods such as using combinatorial libraries or high-throughput screening (HTS) are time-consuming and inefficient. With advancements in computing, methods based on computational techniques or data-driven approaches have emerged to assist in the identification of hit compounds. One approach involves virtual screening techniques from virtual screening libraries containing a large number of molecules, typically utilizing molecular similarity or molecular docking techniques. Another approach involves generating new molecules with specific properties using inverse QSAR, particle swarm optimization, or genetic algorithms, but overall, these methods have not been particularly effective. In today’s artificial intelligence era, the emergence of deep generative models has significantly improved the generation of drug molecules. Generative models can learn the attributes of specific training samples and automatically generate new entities with similar characteristics, making them widely applicable in molecular design, combinatorial optimization, and hit compound discovery. However, existing molecular generative models largely overlook the interactions between ligands and targets. To address this issue, the authors propose a generative model that combines conditional GAN networks and WGAN-GP (WGAN with gradient penalty), linking systems biology and molecular design. This model can generate active molecules using only gene expression features from target gene knockouts, allowing for the generation of hit-like compounds without prior knowledge and demonstrating versatility in designing molecules for multiple targets or biological states within the same model.
2. Model
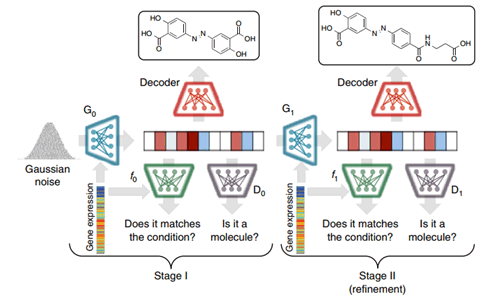
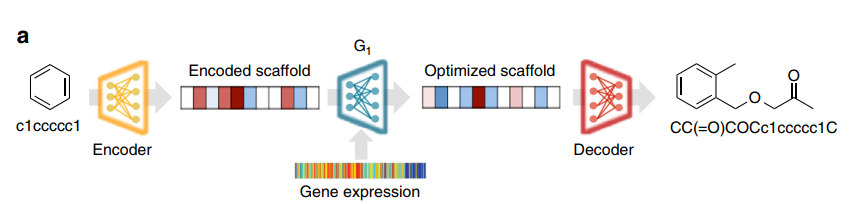
First, the authors use a Grammar VAE model to encode the SMILES sequences of molecules into a latent space, which can then be decoded using a set of grammatical generation rules required to reconstruct the original SMILES sequences. Next, to obtain higher clarity synthetic data, the authors employ StackGAN, which is based on a two-stage progressive approach to molecular generation. The second stage is used to improve the structures generated in the first stage, with the distinction being that the input data for the second stage does not use randomly generated vectors but rather the results generated from the first stage. The structure of the entire model is illustrated as follows:
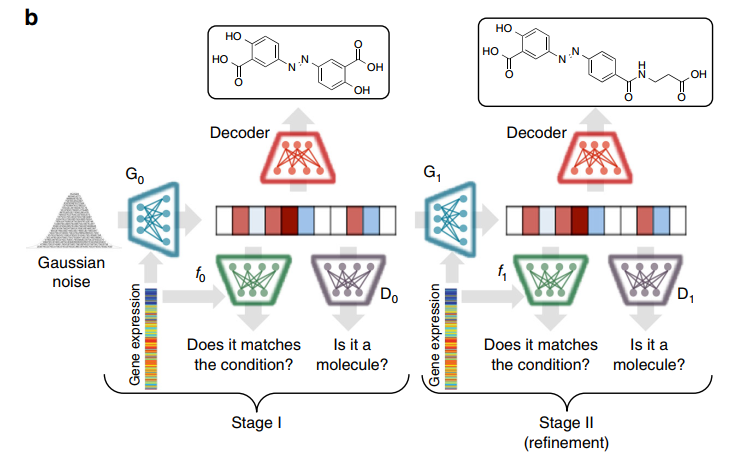
The specific GAN networks used in both stages combine conditional GAN networks and WGAN-GP networks. The reason for employing conditional GAN is to incorporate gene expression features as condition c, while WGAN-GP is an improved model proposed on WGAN. WGAN directly employs weight clipping to handle Lipschitz constraints, but this approach can lead to issues such as gradient vanishing or explosion. Under the combination of the two models, the loss functions for the discriminator and generator in the first stage are as follows:
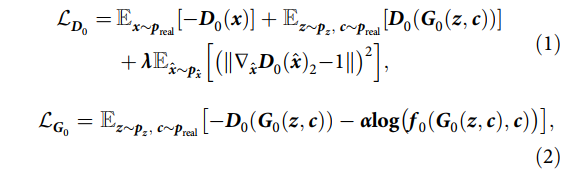
Here, x represents the molecular representation, and c represents the gene expression features used as conditions, both sampled from the true data distribution preal, while the input data z for the first stage follows a Gaussian distribution. The function f0 measures the probability of gene expression features corresponding to the molecular representation, and is a regularization coefficient. The former balances the impact of the gradient penalty term on the discriminator loss, while the latter weights f0, with both set to 10 based on empirical observations. The loss functions for the discriminator and generator in the second stage differ from the first stage as previously explained, and are represented as follows:
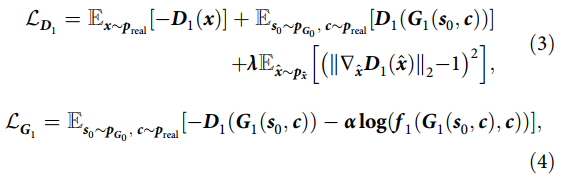
3. Experiments
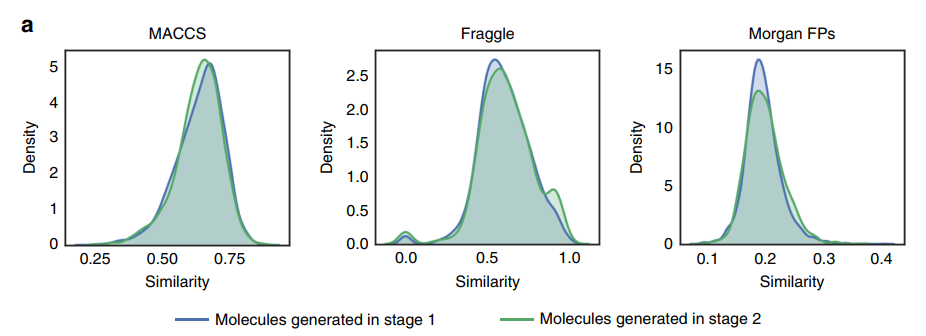
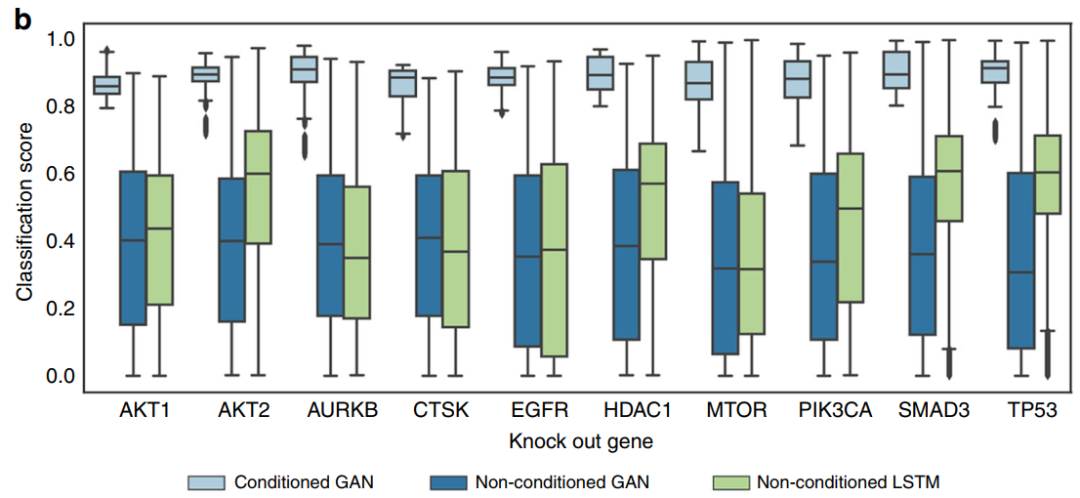
Generating molecules from compound-induced gene expression
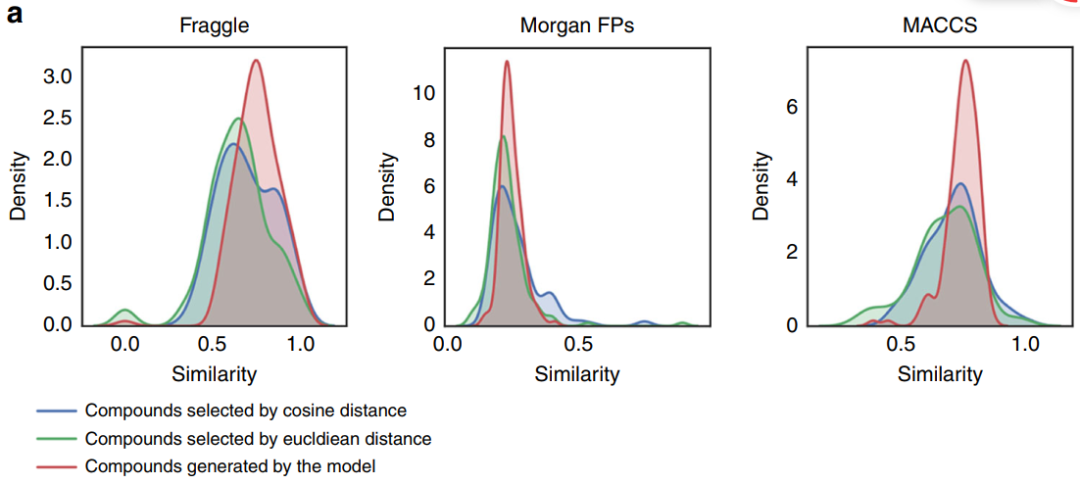
The results indicate that each feature produces approximately 8.5% effective molecules, with about 8.2% being unique SMILES representations and only about 1.6% being easily synthesizable. Additionally, the number of molecules generated in the second stage of StackGAN does not exceed that of the first stage. Furthermore, during molecular generation, the Euclidean distance between reference compounds and the gene expression profiles of the training set can be significant, resulting in reduced similarity to reference compounds. (This is only a portion; please refer to the original text for the rest.)
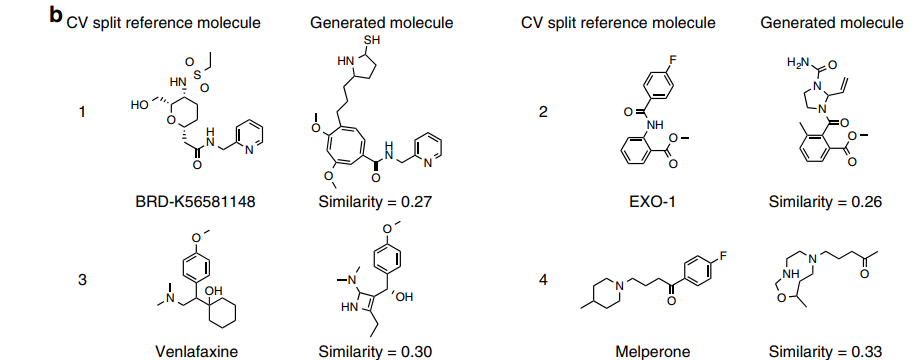
Designing hit-like inhibitor molecules
The figure below displays examples of molecules generated for various targets and their closest known active compounds. (This is only a portion; please refer to the original text for the rest.)
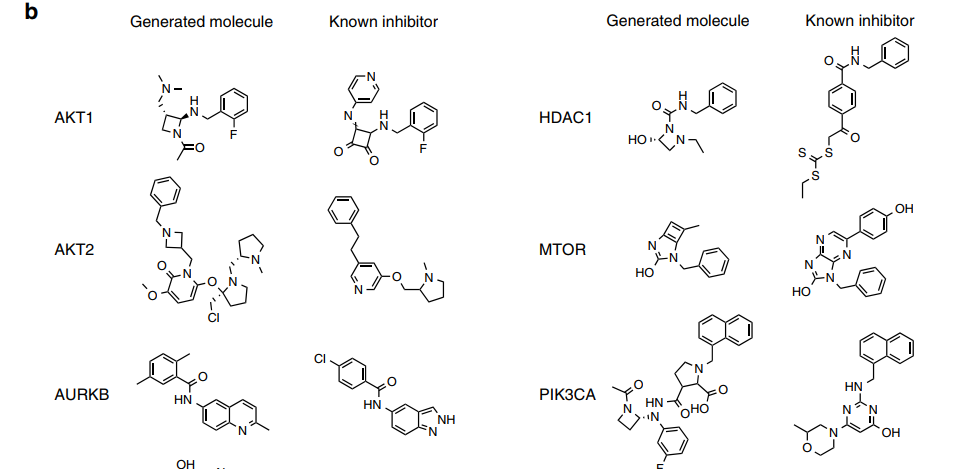
From the above figure, it can be observed that in many cases, the generated molecules share functional groups with active compounds, and even have similar long sequence fragments. These examples demonstrate that the gene expression features of targets post-gene knockout can guide the generation of molecules towards specific regions of chemical space associated with active compounds.
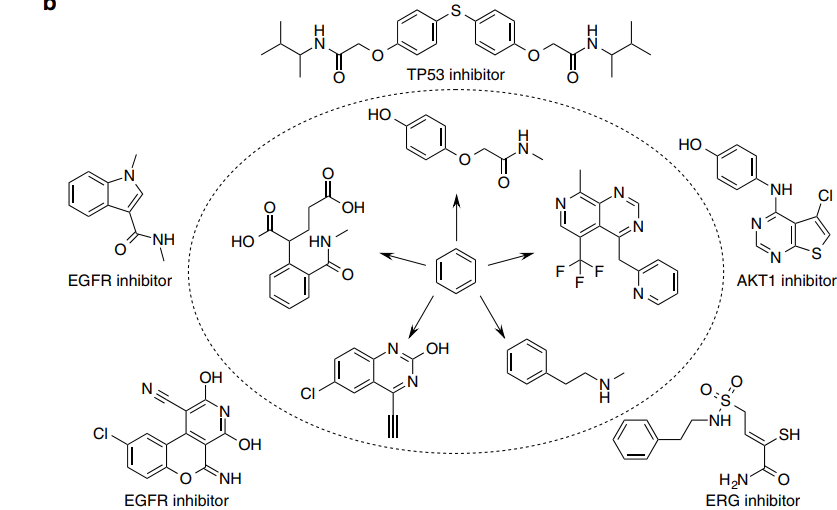
Optimizing long sequence fragments (scaffolds)
The figure below presents examples of molecules optimized based on specific targets, generated by optimizing the phenyl ring after gene knockout of AKT1, EGFR, ERG, and TP53, with the most relevant and nearest active compounds displayed outside the circles.
Similarity search and comparison with the model
Specific regions of chemical space
4. Conclusion
The authors propose a method based on conditional generative adversarial networks that generates new molecules from specific gene expression features, making it highly useful in situations where target annotations and activity data are unavailable, such as in target de-isolation. However, there is still room for improvement in this method, such as assessing whether it can be applied to the optimization of lead compounds or finding methods to generate compounds with known structural features related to specific drug target activities. The authors also plan to expand this method to automatically generate molecules with multi-target features or capable of reversing toxicological or disease-related gene expression features.
References
Méndez-Lucio, O., Baillif, B., Clevert, D. et al. De novo generation of hit-like molecules from gene expression signatures using artificial intelligence. Nat Commun 11, 10 (2020). https://doi.org/10.1038/s41467-019-13807-w
Code: Please contact the relevant authors for access.
References