Advancements in AI Technology for Lead Compound Discovery and Optimization

Source

The Journal of Pharmaceutical Sciences 2024, 59(9): 2443−2453

Authors

Li Ziyue, Cong Kaiyuan, Wu Shiqi, Zhu Qihua, Xu Yungen, Zou Yi
School of Pharmacy, China Pharmaceutical University

Abstract

In recent years, the development of artificial intelligence (AI) technology has made significant progress and has been widely applied in various fields such as medicine and pharmacy, accelerating the drug development process. This article focuses on the application of AI in the discovery and optimization of lead compounds, detailing AI-assisted virtual screening and molecular generation methods for discovering lead compounds, especially cases of AI-driven drugs entering clinical trials. It also briefly discusses the application of basic AI algorithm models in quantitative structure-activity relationship (QSAR) and drug repositioning, providing references for AI-based drug discovery.

Keywords

artificial intelligence; drug discovery; lead compounds; virtual screening; molecular generation
|
Main Text |
Artificial intelligence (AI) is a science that uses computer technology, theories, methods, and software to study and develop systems that can expand human intelligence, simulate human behavior and thinking, and analyze data. It mainly includes two subfields: machine learning (ML) and deep learning (DL). With the rapid development of computer technology and the advent of the big data era, AI has achieved breakthroughs in various fields, such as AlphaGo defeating professional Go players using DL technology and the recent popularity of chatbot programs ChatGPT and text-to-video generation models Sora, which have made significant impacts in various areas.
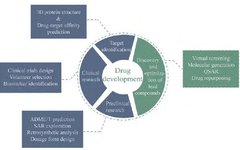
With the rapid development and promotion of AI technology, AI+ has permeated various aspects of the healthcare field. Initially, AI was widely applied in medical imaging, and gradually penetrated the field of drug development, leading to a series of changes in drug development models. In recent years, as biomedical data continues to accumulate, the application of AI technology in pharmacy has become increasingly widespread. The data generated during the process of new drug development involves all stages from target discovery, lead compound discovery and optimization, to preclinical research and clinical trials. AI can quickly mine high-density information from massive raw data in drug development and provide new insights for drug development by integrating and analyzing these data. Today, AI technology is gradually participating in all stages of the drug discovery process, with academia and industry attempting to study the use of AI to assist drug development, seeking support for the discovery and development of new drugs (Figure 1).
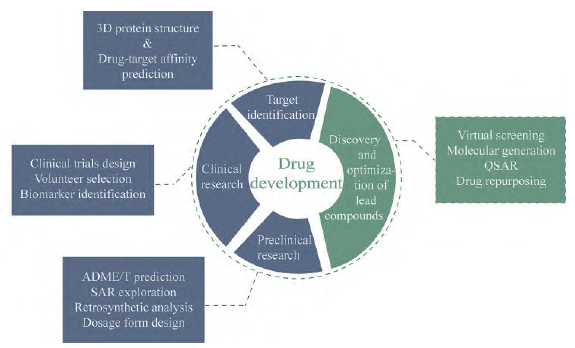 Figure 1 Application of artificial intelligence technology in the drug development process. QSAR: Quantitative structure-activity relationship; ADME/T: Absorption, distribution, metabolism, excretion/toxicity; SAR: Structure-activity relationship
Figure 1 Application of artificial intelligence technology in the drug development process. QSAR: Quantitative structure-activity relationship; ADME/T: Absorption, distribution, metabolism, excretion/toxicity; SAR: Structure-activity relationship
Drug discovery is a long and complex process, among which the discovery and optimization of lead compounds is a core step in new drug development, requiring a repeated cycle of “design, synthesis, and activity testing” to improve the activity, selectivity, and drug-likeness of compounds. Currently, the discovery and optimization process of lead compounds heavily depends on the experience of medicinal chemists and substantial resource investment, resulting in slow speeds and high workloads. However, under the drive of AI, the discovery and optimization of lead compounds are expected to balance speed and accuracy, providing opportunities to enhance efficiency and reduce costs. This article will focus on the application of AI in the discovery and optimization process of lead compounds, briefly introducing the basic concepts and algorithms of machine learning and deep learning, and then emphasizing specific cases of ML/DL algorithms in the discovery and optimization of lead compounds to illustrate the important role of AI in the drug design process.
1
Introduction to Concepts and Algorithms of Machine Learning and Deep Learning
Machine learning is a way to achieve artificial intelligence and is a subfield of AI. Machine learning automatically identifies and analyzes data based on existing data, knowledge, or experience, summarizes meaningful patterns, and makes predictions or decisions in similar environments. It is mainly divided into two categories: supervised learning and unsupervised learning. Supervised learning refers to learning or building a model from a labeled training set and predicting new instances based on that model. In contrast, models in unsupervised learning use unlabeled datasets for training and allow operations on that data without any supervision.
1.1 Introduction to Common Machine Learning Algorithms
1.1.1 Naive Bayes (NB)
NB is a common classification method whose core idea is to predict categories by considering feature probabilities, i.e., for a given sample to be classified, solving the probability of each category occurring under the condition of this sample appearing.
1.1.2 K-Nearest Neighbors (KNN)
The KNN algorithm, proposed by Cover and Hart in 1967, is a classification algorithm. Its overall idea is that the neighbors of a sample are labeled samples, and for a sample X to be classified, the category with the most samples in the closest neighborhood will classify that sample into the same category (Figure 2A).
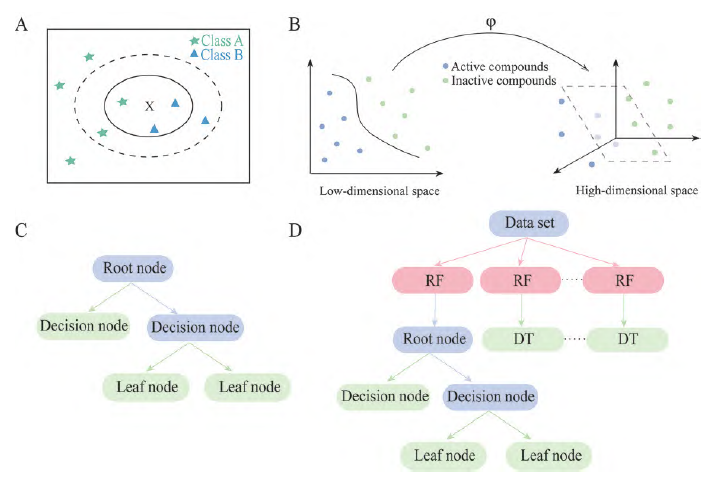 Figure 2 Introduction to machine learning algorithms. A: The basic principle of KNN algorithm; B: SVM maps low-dimensional data to high-dimensional space for separation; C: The diagram of DT algorithm model; D: The diagram of RF algorithm model. KNN: K-nearest neighbors; SVM: Support vector machines; DT: Decision tree; RF: Random forest
Figure 2 Introduction to machine learning algorithms. A: The basic principle of KNN algorithm; B: SVM maps low-dimensional data to high-dimensional space for separation; C: The diagram of DT algorithm model; D: The diagram of RF algorithm model. KNN: K-nearest neighbors; SVM: Support vector machines; DT: Decision tree; RF: Random forest
1.1.3 Support Vector Machines (SVM)
SVM, proposed by Cortes et al. in 1995, is often used for classification problems. The idea of SVM is that training data in its original low-dimensional input space can be separated in a high-dimensional latent space constructed through mapping (Figure 2B).
1.1.4 Decision Trees (DT) and Random Forests (RF)
DT is a commonly used classification algorithm that analyzes data by constructing a tree-like decision structure based on known feature values. The decision tree model is characterized by readability and fast classification speed, making it widely used in various practical business modeling processes (Figure 2C).
RF is a classifier that trains and predicts samples by constructing multiple DTs, where the category with the most classifications from DTs is taken as the final result (Figure 2D).
1.1.5 Extreme Gradient Boosting (XGBoost)
XGBoost is an ensemble learning algorithm based on decision trees that iteratively trains weak learners, typically decision trees, and adjusts the model based on errors from the previous iteration, gradually improving the overall model’s performance. XGBoost is a powerful, flexible, and efficient machine learning algorithm applicable in various fields and problems.
1.2 Introduction to Concepts and Models of Deep Learning
Deep learning is a new field in machine learning research that utilizes deep artificial neural networks to automatically learn, classify, and predict from vast amounts of data, identifying features within the data.
1.2.1 Deep Neural Networks (DNN)
Artificial neural networks are computational models inspired by the biological neural networks of the human brain, excelling at learning mapping relationships from input data to solve prediction or classification problems. They are similar to biological neural networks and consist of artificial neurons. Each neuron simulates the signal transmission and activation of biological nerve cells through simple mathematical models. It is generally believed that a neural network with more than three layers can be called a deep neural network. Feedforward refers to the one-way propagation direction of the neural network, where signals propagate in one direction. Although the feedforward network structure is simple, it cannot handle time series data well, leading to the emergence of recurrent neural networks.
1.2.2 Recurrent Neural Networks (RNN)
RNN, proposed by Hopfield in 1982, refers to a fully connected neural network that incorporates relationships in time sequences, allowing it to better handle time-related problems such as machine translation.
Traditional neural network models consist of an input layer, hidden layers, and an output layer, with full connections between layers, while nodes between layers are unconnected. This ordinary neural network struggles with time series data. Compared to feedforward neural networks, RNNs can receive the hidden state from the previous time point, indicating that the network state at the previous moment will influence the next moment’s network state, showing the close relationship between RNNs and sequential data (Figure 3A).
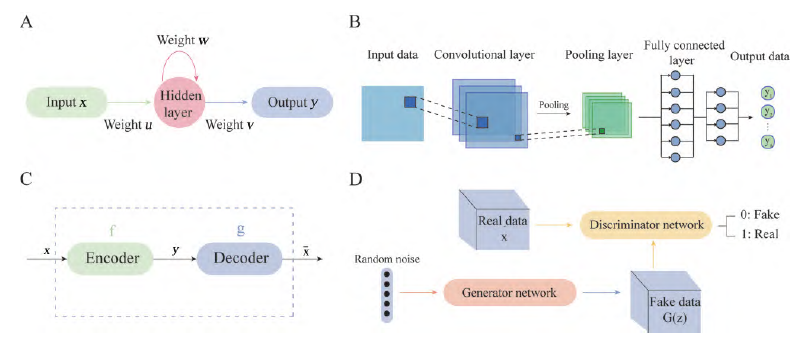
Figure 3 Introduction to deep learning algorithms. A: The structure of RNN; B: The structure of CNN; C: The components of AE; D: The mechanism of GAN model. RNN: Recurrent neural networks; CNN: Convolutional neural networks; AE: Autoencoder; GAN: Generative adversarial networks
Long Short-Term Memory networks (LSTM) are a special type of RNN particularly suitable for processing sequential data. LSTM was proposed by Hochreiter and Schmidhuber in 1997, specifically designed to avoid the long-term dependency problem of RNNs. It has been successfully applied in various fields such as speech recognition, image description, and natural language processing.
In 2020, Yang et al. used an LSTM-based neural network model to train two hundred thousand compounds from the ChEMBL database. They then fine-tuned the model using a dataset containing 135 published E1A-binding protein inhibitors and 576 macrocyclic molecules to generate new p300/CREB-binding protein inhibitors. The model generated a concentrated library of 672 chemical structures, from which some compounds were selected for synthesis. After further systematic optimization, an efficient inhibitor was obtained. Among them, the potential candidate drug B026 exhibited high inhibitory activity against p300/CBP histone acetyltransferase in human cancer animal models and showed significant tumor growth inhibition, and has been identified for further preclinical development.
In 2022, Li et al. proposed a generative deep learning (GDL) model, which is based on an LSTM algorithm and a distributed learning recurrent neural network architecture capable of generating new molecules that follow the same chemical distribution as the training set. Its role is to generate a customized virtual compound library for a given biological target. Subsequently, the GDL model was applied to screen receptor-interacting protein kinase 1 (RIPK1) kinase inhibitors, successfully discovering a novel scaffold RIPK1 kinase inhibitor, demonstrating the important role of deep neural networks in early drug discovery.
1.2.3 Convolutional Neural Networks (CNN)
CNN is a type of feedforward neural network that includes convolutional calculations and has a deep structure, consisting of input layer, convolutional layer, activation function, pooling layer, and fully connected layer. The core of CNN is the convolutional layer, which can model specific prediction problems, learning a large number of convolutional kernels from the training dataset. After convolutional calculations, a rectified linear unit (ReLU) function is often added to non-linearize the data. The pooling layer effectively reduces the size of the matrix, speeding up computation and preventing overfitting. Finally, a fully connected layer is used for final classification (Figure 3B).
In 2022, Noguchi et al. proposed a Pixel Convolutional Neural Network (PixelCNN) model that converts simplified molecular input line entry system (SMILES) strings into two-dimensional matrix data, applying masked neural network layers to build the model. They conducted multifaceted analyses of PixelCNN’s performance and made detailed comparisons with RNNs in generating expected molecular properties and in exploring chemical space based on fragment growth optimization. Despite RNNs outperforming PixelCNN in directly predicting molecular structures corresponding to target molecular properties, the PixelCNN-based framework significantly excelled in fragment growth optimization of molecular structures, making it well-suited for fragment-based drug discovery tasks.
1.2.4 Autoencoders (AE)
AE, as a method of deep neural networks, is mainly used for dimensionality reduction, compression, and obtaining low-dimensional representations. Autoencoders serve the same purpose as traditional dimensionality reduction methods such as principal component analysis, but are often more flexible and effective compared to them.
Autoencoders can learn the implicit features of input data, called encoding, while reconstructing the original input data using the learned new features, called decoding. The main goal is to transform the input data x into an intermediate variable y, and then transform y back into x̄, comparing input x and output x̄ to make them as close as possible (Figure 3C).
In 2020, Chenthamarakshan et al. proposed a generative model called Controlled Generation of Molecules (CogMol), which introduced a multi-attribute controlled sampling scheme into the variational autoencoder model to design a new set of molecules with desired attributes for viral proteins. They used CogMol to generate new molecules for three target proteins of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), constraining their target affinity and selectivity, drug similarity, synthetic feasibility, and toxicity. The results showed that the generated molecules could bind favorably to the target-related drug pockets and exhibited low predicted metabolite toxicity and high synthetic feasibility.
1.2.5 Generative Adversarial Networks (GAN)
GAN is a type of generative model that trains models by acquiring samples and then generating data according to defined target data distributions (Figure 3D). GAN consists of a generator and a discriminator; the generator captures the latent distribution of real data samples and generates new data samples, while the discriminator is a binary classifier that distinguishes between real data and generated samples. The generator and discriminator continuously optimize to enhance their respective generation and discrimination capabilities, and this learning optimization process seeks to find a Nash equilibrium between the two.
In 2018, Polykovskiy et al. proposed an Entangled Conditional Adversarial Autoencoder (ECAAE) model that generates molecular structures based on various attributes, such as activity against specific proteins, solubility, or synthetic performance. Researchers used ECAAE to generate a new Janus kinase 3 (JAK3) inhibitor, and the discovered molecule showed good activity and selectivity in in vitro tests.
The content of this article was selected by the editor of Fanmo Valley from the literature, and the formatting and editing are original. If reproduced, please respect the labor results and indicate [Source: Fanmo Valley WeChat Official Account].
2
Application of AI Technology in Lead Compound Discovery and Optimization
2.1 Virtual Screening
Virtual screening refers to the pre-screening of compound molecules on a computer based on predetermined conditions before conducting biological activity screening, to identify the small molecules most likely to bind to the target, thereby significantly reducing the number of compounds that need to be screened in practice while improving the efficiency of discovering lead compounds. Virtual screening has long been one of the powerful means for discovering hit and lead compounds. However, the drawback of virtual screening is that as the scale of the virtual compound library expands, its screening speed and efficiency are relatively low; for virtual screening strategies based on molecular docking, the accuracy of their scoring functions needs improvement. In recent years, applying various AI algorithms to virtual screening can expand the number and diversity of molecules in the virtual compound library on one hand, and improve screening algorithms and develop AI-based scoring functions to make screening results more accurate on the other hand.
2.1.1 Large-Scale Virtual Screening
The commercial compound libraries for virtual screening generally contain fewer than ten million available compounds, which is only a small fraction compared to the potential 1060 drug-like compound space. This limitation reduces the efficiency of drug discovery through virtual screening. Later, virtual compound libraries at the billion-level were developed, such as the Enamine company constructing virtual libraries through molecular building blocks and chemical reaction combinations, with compound numbers reaching the hundred billion level. However, as the scale of virtual libraries increases to the billion level, the efficiency of screening using traditional virtual screening algorithms becomes limited, necessitating the development of efficient and rapid methods for large-scale compound library virtual screening.
In 2021, Kalliokoski developed software called macHine leArning booSTEd dockiNg, or HASTEN, which is the first tool evaluated on billion-level benchmarks. By iteratively training machine learning models, it predicts molecular docking scores to accelerate structure-based virtual screening. They subsequently used the HASTEN tool to screen 1.56 billion molecules from the Enamine REAL class lead compound virtual library in cases of antibacterial molecules and antiviral kinases: first randomly selecting 0.1% of virtual compounds as starting data for conventional molecular docking, training the machine learning model to predict docking scores for all molecules in the compound library and ranking them, continuing to select the top 0.1% of compounds for molecular docking, repeating this process nine times, and ultimately the total training dataset reached 1% of the entire virtual library. The results showed that when docking only 1% of the compounds in the library, HASTEN could identify a thousand true high-scoring virtual hits with a high recall rate of 90%. Thus, HASTEN is an effective strategy suitable for screening billion-level libraries in daily drug discovery.
In the same year, Sadybekov et al. published a method called virtual synthon hierarchical enumeration screening (V-SYNTHES) in Nature, which can perform hierarchical screening on virtual libraries of over 11 billion compounds, significantly reducing the number of molecules that need to be evaluated when searching for potential hit compounds, using only 1/100 of the computational resources compared to standard methods. This method can be easily scaled to accommodate the rapid growth of combinatorial libraries and may be applicable to any docking algorithm. They later tested the screening capabilities of V-SYNTHES in Rho-associated protein kinase 1 (ROCK1), and after synthesizing and testing 21 hit compounds, found that 6 compounds could bind to ROCK1 kinase, with inhibition constants Ki values less than 10 μmol·L-1.
In 2022, Beroza et al. proposed a virtual screening method that stores potential products as building blocks (molecular fragments) and dynamic generation rules for compounds, then uses a feature tree algorithm to search for building blocks and compare them, connecting the best building blocks with complementary ones based on chemical rules while considering synthetic feasibility to generate new molecules. This method expands structure-based fragment assessment to a broader chemical space, subsequently applying this method to screen ROCK1 inhibitors from nearly one billion virtual compounds, selecting 69 molecules for purchase and validation, of which 27 had Ki values less than 10 μmol·L-1, achieving a hit rate of up to 39%. Moreover, this docking method is several orders of magnitude faster than traditional docking methods.
In 2023, Gryniukova et al. applied a new AI-based method called PharmAI DiscoveryEngine to perform virtual screening on a compound library containing 2.6 million molecules, identifying 434 potential small molecules targeting silent information regulator 1 (Sirtuin-1), accounting for only 0.02% of the entire library. After multi-stage in vitro validation, they successfully discovered 9 novel Sirtuin-1 inhibitors with unique chemical structures. Finally, using liquid chromatography/mass spectrometry (LC/MS) untargeted methods to test the inhibitory activity of the compounds, the results showed that two compounds exhibited the best inhibitory activity (Figure 4). This indicates that AI tools can reduce large-scale screening datasets to only a few hundred small molecule compounds targeting specific targets, making the discovery process for lead compounds faster and more economical.
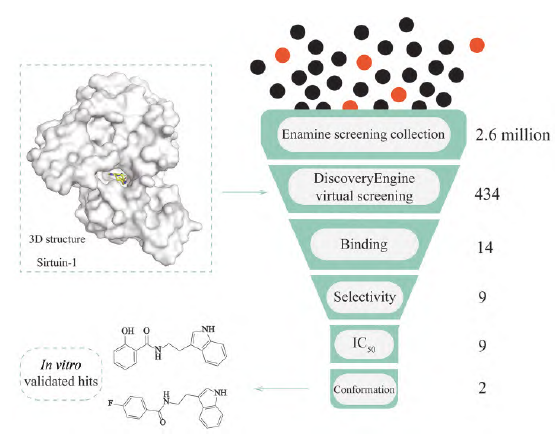
Figure 4 Artificial intelligence (AI)-assisted virtual screening process for novel Sirtuin-1 small molecule inhibitors. Sirtuin-1: Silent information regulator 1
2.1.2 Virtual Screening Scoring Functions
To efficiently obtain highly active molecules, in addition to needing rapid screening computational methods, developing more reliable scoring functions is particularly important for virtual screening based on molecular docking.
In 2020, Professor Hou Tingjun’s research group at Zhejiang University proposed a scoring function named EAT-Score (energy auxiliary terms score) by combining the XGBoost method in machine learning with energy auxiliary terms in molecular docking scoring, including five different classical docking scoring energy scores and protein-ligand interaction fingerprint terms (Figure 5). Validation with the DUD-E (database of useful decoys-enhanced) dataset showed that EAT-Score significantly outperformed classical scoring functions in virtual screening, with the area under the receiver operating characteristic (ROC) curve increasing by approximately 0.3 compared to traditional methods.
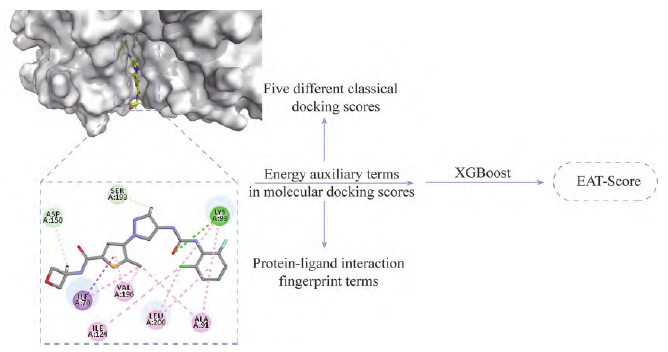
Figure 5 The design process of EAT-Score. XGBoost: Extreme gradient boosting; EAT-Score: Energy auxiliary terms score
In 2022, Morris et al. created a machine learning consensus docking tool (MILCDock), which adopts a multilayer perceptron approach to integrate the binding affinities and binding mode predictions of five traditional molecular docking tools, assigning a comprehensive score, with larger scores indicating higher binding affinity. Testing on the DUD-E and LIT-PCBA (a bias-free dataset for machine learning and virtual screening) datasets showed that MILCDock outperformed traditional molecular docking tools and other consensus docking methods on the DUD-E dataset. This demonstrates that MILCDock not only integrates the advantages of traditional tools but also achieves higher predictive accuracy through machine learning models.
In 2023, Zhang et al. proposed a new molecular docking scoring function called theory-based interaction energy component score (TB-IECS), which combines energy terms from Smina and NNScore2 (neural-network scoring function version 2) and constructs a model using XGBoost. TB-IECS outperformed traditional scoring functions like Glide SP (standard precision Glide docking) and Dock in the DUD-E dataset, LIT-PCBA dataset, and practical virtual screening scenarios, effectively balancing efficiency and accuracy. TB-IECS has shown superiority among machine learning scoring functions, promising to become an accurate virtual screening method.
2.1.3 AI-Assisted Virtual Screening for Clinical Candidate Compounds
Pharos iBio is a new drug development company specializing in treatments for rare and refractory diseases, utilizing its big data and AI technology-based new drug development platform, Chemiverse, to develop new drugs (Figure 6A). The Chemiverse platform supports the use of various algorithms, 10 different modules, and over 230 million big data entries, covering multiple aspects from target identification, lead compound discovery and optimization to clinical stages, thereby accelerating drug discovery and shortening research and development cycles. Pharos iBio has developed several new drugs using this platform, one of which is the next-generation FMS-like tyrosine kinase 3 (FLT3) inhibitor PHI-101. In preclinical studies, PHI-101 exhibited higher anti-leukemia activity and better selectivity compared to marketed FLT3 inhibitors midostaurin and gilteritinib, while also showing lower toxicity. PHI-101 is currently undergoing two Phase I clinical trials for acute myeloid leukemia and high-grade serous ovarian cancer (NCT04842370/NCT04678102).
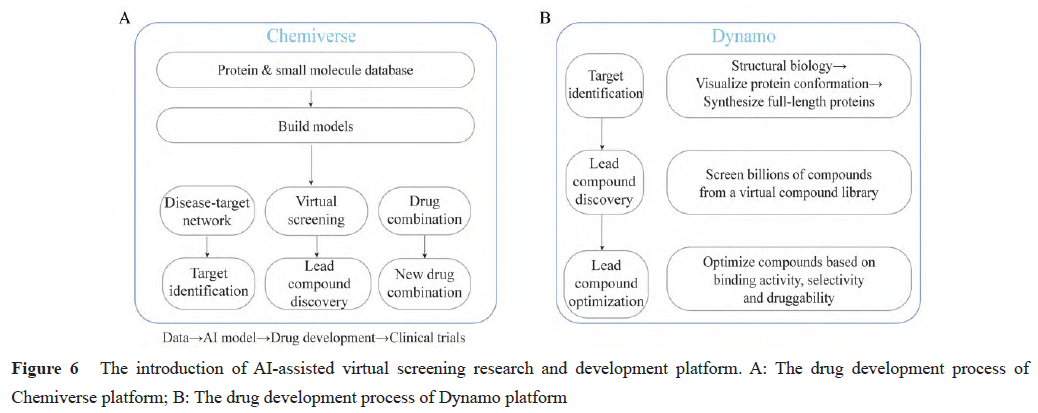
Relay Therapeutics is a precision therapy company targeting small molecule drug discovery for tumors. The company integrates cutting-edge experimental and computational methods to establish the Dynamo platform. The Dynamo platform is divided into three modules: drug target identification, lead compound discovery, and lead compound optimization (Figure 6B). The Dynamo platform first inputs the structural biology information of proteins into the Anton2 computing platform to generate virtual simulations of full-length proteins over long periods, aiding in better understanding protein dynamics and identifying drug targets. It then uses cloud computing to perform virtual screening on billions of molecules from the virtual compound library to obtain a large number of structurally novel small molecules with potential activity. The Dynamo platform can also optimize lead compounds based on efficacy, selectivity, bioavailability, and drug-likeness indicators, and conduct compound synthesis and activity testing in the laboratory; the wet lab results obtained are then compared with computational predictions to train machine learning models, thereby improving computational prediction models. Currently, Relay Therapeutics has developed four clinical candidate drugs through this platform: fibroblast growth factor receptor 2 (FGFR2) inhibitor RLY-4008 (Phase I, NCT04526106), Src homology-2-containing protein tyrosine phosphatase 2 (SHP2) inhibitor RLY-1971 (Phase I, NCT04252339), phosphoinositide 3-kinase alpha (PI3Kα) inhibitor RLY-2608 (Phase I, NCT05216432), and RLY-5836 (Phase I, NCT05759949).
2.2 Molecular Generation
In recent years, deep learning has been widely used in molecular generation, enabling de novo drug design to obtain entirely new scaffolds for drug molecules and improve the novelty of drug molecules.
In 2021, Professor Lai Luhua’s research group at Peking University developed a novel drug design method called DeepLigBuilder, which uses deep generative models to directly construct and optimize ligands within target binding pockets. First, a model capable of generating drug-like molecules with effective three-dimensional structures is trained, and then information based on the target is introduced into the model to obtain molecules with good predicted binding strength. They used DeepLigBuilder to design potential inhibitors for the main protease (Mpro) of SARS-CoV-2, with experimental results showing that DeepLigBuilder discovered compounds with new interactions with the target protein, though further experimental verification is needed to determine if these compounds effectively inhibit Mpro.
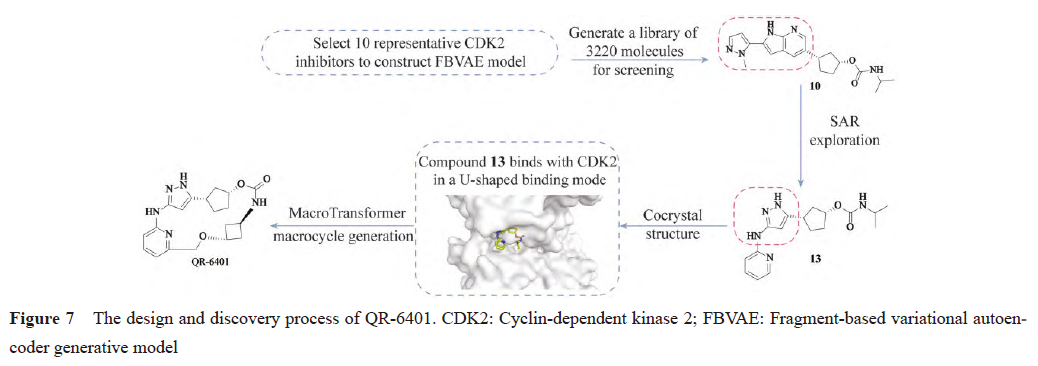
In 2023, Yu et al. discovered an efficient and highly selective cyclin-dependent kinase 2 (CDK2) inhibitor QR-6401 by applying generative models and structure-based drug design. They first developed a fragment-based variational autoencoder generative model (FBVAE) targeting 10 published CDK2 inhibitors, generating a library of 3220 molecules and screening to obtain 10 compounds through Glide Docking. Through structural modifications, they obtained compound 13, which exhibited certain activity and selectivity, with the crystal structure showing a U-shaped binding between CDK2 and compound 13. They then derived compound 13 into the most active and selective compound QR-6401 using the MacroTransformer method, which demonstrated good anti-tumor efficacy in the OVCAR3 (human ovarian cancer) xenograft model. Notably, the Shanghai Institute of Materia Medica of the Chinese Academy of Sciences recently reported a deep generative method called D3Rings that can quickly identify and generate cyclic compounds, providing important support for drug discovery and development.
Currently, many large AI pharmaceutical companies both domestically and internationally have developed their molecular generation platforms to accelerate the discovery and optimization process of lead compounds. In 2017, Insilico Medicine trained an adversarial autoencoder using publicly available data from the American Cancer Society, which can generate candidate molecules with potential anti-cancer properties based on target molecular features. In this study, Insilico Medicine predicted 69 compounds and synthesized and tested four of them, with results indicating that the predicted compounds exhibited good activity. In 2019, the Insilico Medicine team proposed a generative tensorial reinforcement learning (GENTRL) model, training it on the ZINC molecular dataset, and using publicly available kinase inhibitor datasets to generate 40 candidate molecules targeting discoidin domain receptor 1 (DDR1), from which six compounds were selected for synthesis and validation. In vitro experiments showed that four of these compounds had good enzyme inhibitory activity against DDR1, with IC50 values reaching 10 nmol·L-1. This time-efficient study validated the excellent predictive performance of the GENTRL model. In 2020, the research team released an AI molecular generation tool called Chemistry42, which integrates various cutting-edge algorithm models, including generative autoencoders, generative adversarial networks, flow-based generative models, evolutionary algorithms, and language models. The main advantage of this platform is its personalized reward mechanism; Chemistry42 continuously evaluates the generated molecular structures using reward mechanisms and three-dimensional physical structure modules, conducting multi-dimensional scoring and optimization covering efficacy, synthetic difficulty, pharmacokinetic properties, etc.
Between 2021 and 2023, Insilico Medicine has multiple internally developed projects in clinical stages, involving fibrosis, tumors, and antiviral areas, further validating the capabilities of its AI molecular generation platform. Among them, the most notable candidate drug ISM001-055 for treating idiopathic pulmonary fibrosis entered Phase I clinical trials (NCT05154240) in less than 30 months, using only a fraction of the traditional drug development costs. In February 2023, ISM001-055 received FDA orphan drug designation and is currently in Phase II clinical trials (NCT05975983). Notably, this is the world’s first drug entirely designed by AI to enter Phase II clinical trials. Additionally, Insilico Medicine has also made significant breakthroughs in identifying novel lead compounds by combining its generative AI platform with AlphaFold, particularly in the fields of cyclin-dependent kinase 20 (CDK20) inhibitors and salt-inducible kinase 2 (SIK2) inhibitors.
2.3 Other Applications of AI Technology
2.3.1 Quantitative Structure-Activity Relationship (QSAR)
In 1962, Hansch made pioneering work on QSAR, which is an important component of drug discovery that can efficiently and cost-effectively predict molecular activity and properties. Classical QSAR methods mainly establish qualitative/quantitative relationships between various descriptors and biological activity through mathematical models. Descriptors include molecular fingerprints, graphs, or other mathematical representations; biological activity includes enzyme activity, affinity, binding free energy, pharmacokinetic properties/toxicity (ADME/T), etc. Due to the widespread use of mathematical models, QSAR has early combined with machine learning algorithms, with two of the most successful machine learning algorithms applied to QSAR being RF and DNN.
In 2019, Shi et al. established a CNN model based on two-dimensional molecular images, effectively predicting pharmacokinetics and toxicity properties of drug molecules, including human cytochrome P450 enzyme 1A2 (CYP1A2) and P-glycoprotein inhibitory activities, blood-brain barrier permeability, and Ames mutagenicity. The results indicated that the established CNN model’s predictive ability is comparable to existing machine learning models based on manual structure descriptors and feature selection, demonstrating that CNN can effectively extract key image features related to molecular ADME/T properties and provide useful tools for virtual screening and drug design research.
In 2020, Arian et al. used the KNN model to identify active protein kinase inhibitors and employed genetic algorithms to extract optimal descriptors. To evaluate the performance of the proposed model, SVM and NB models were used for testing, with results showing that the KNN model’s outputs significantly outperformed other QSAR models.
In 2021, Zhou et al. collected 1785 potential human immunodeficiency virus 1 (HIV-1) inhibitors, randomly sampling to divide the database into training and testing sets. Based on the training set, a classifier for HIV-1 inhibitors was established using the NB model. Validation with the testing set showed that the NB model predicted 88.3% of inhibitors and 87.2% of non-inhibitors, with a correlation coefficient of 85.2%, also obtaining key molecular fragments.
In 2021, Kumar et al. developed a method called B3Pred to predict blood-brain barrier penetrating peptides (B3PPs). The developed model was trained, tested, and evaluated on blood-brain barrier peptides obtained from the B3Pdb (database of blood-brain barrier crossing peptides), where the RF-based model performed best among the top 80 selected features, achieving a maximum accuracy of 85.08%, with an area under the receiver operating characteristic (AUROC) value of 0.93. B3Pred is now available on the web, including three main modules for predicting, designing B3PPs, and scanning for B3PPs in protein sequences.
In 2022, Wu et al. established a hierarchical support vector regression (HSVR) model based on machine learning to predict skin permeability coefficients and reveal intrinsic permeation mechanisms. The established HSVR model showed excellent performance across training, testing, and outlier datasets, with various statistical assessments and validation confirming the model’s accuracy and predictability.
Over the past few decades, QSAR has become an indispensable tool in the drug development processes of major pharmaceutical companies. The integration of QSAR and machine learning methods has transformed the concept of drug discovery from rule-driven to data-driven, facilitating the discovery of new compounds. However, machine learning methods rely on experimental data, and due to the lack of standardization in experimental conditions, experimental data is often imbalanced and noisy. Future efforts should focus on establishing large but consistent experimental datasets and developing new machine learning algorithms.
2.3.2 Drug Repositioning
Drug repositioning, also known as