

The complement system is a component of innate immunity. It plays an important role in defending against pathogen infections, participating in immune regulation, and maintaining internal environmental stability; however, abnormal activation of the complement system can also cause self-damage, leading to diseases. The kidneys are the organs most susceptible to damage. In recent years, the mechanisms of complement involvement in various kidney diseases have gradually been elucidated, and drugs targeting different stages of complement activation have emerged, some of which have been applied in clinical practice. However, there are many issues regarding complement therapy. In light of this, Peking University Health Science Center organized experts from nephrology, public health, and preventive medicine to comprehensively search for literature related to complement-related kidney disease (CMKD), including PubMed, Elsevier Science Direct, Wiley Online Library, Wanfang Database, and China National Knowledge Infrastructure. They also supplemented the search with domestic and international guidelines, books, and recommendations related to kidney diseases, mainly including the official websites of the European and American Societies of Nephrology and notifications from the National Health and Family Planning Commission. The types of literature included guidelines, expert consensus, systematic reviews, randomized controlled trials, cohort studies, case series studies, etc. The search was completed by July 2023. Based on domestic and international guidelines/consensus on kidney diseases and the latest evidence, after three rounds of expert discussion meetings, this consensus was finally formed to provide reference for the diagnosis and treatment of CMKD.
1. Complement System
(1) Introduction
The complement system consists of more than 50 plasma proteins and membrane proteins, including complement inherent proteins, complement receptors, and complement regulatory proteins. Complement activation includes three pathways: the classical pathway, the lectin pathway, and the alternative pathway. The classical pathway is primarily activated by immune complexes formed by antigens and antibodies; the lectin pathway is mainly activated by mannose-binding lectin (MBL) recognizing pathogens or mannose sites exposed by self; under physiological conditions, the alternative pathway is spontaneously activated through continuous hydrolysis of C3. C3 convertase (C4b2a or C3bBb) cleaves C3 to generate C3b and C3a; C3b binds to C3 convertase to form C5 convertase, cleaving C5 to generate C5a and forming the membrane attack complex (MAC). Complement regulatory proteins participate in regulating complement activation to avoid excessive complement activation that causes host tissue damage. Complement factor I cleaves C3b/C4b with the assistance of complement factor H, complement receptor 1, membrane cofactor protein (MCP, CD46), and C4-binding protein; complement factor H can inhibit and degrade C3 convertase. Decay-accelerating factor (DAF, CD55) is a membrane surface regulatory protein that prevents the formation of C3 convertase. MAC inhibitory factor CD59 prevents C9 from inserting and polymerizing on the cell membrane. Collectins and ficolins capture and clear soluble precursors of MAC, inhibiting the complement cascade reaction.
After complement activation, a series of effector molecules are generated, which have a wide range of biological functions. C3b (and its degradation products iC3b and C3dg) and C4b deposit on the surface of pathogens or damaged cells and can interact with complement receptors on the surface of phagocytes to assist phagocytes in clearing pathogens and apoptotic host cells; complement activation generates large amounts of C3a and C5a, which interact with specific receptors on various cells (such as granulocytes, dendritic cells, macrophages, as well as endothelial cells and epithelial cells) (C3aR, C5aR1, C5aR2), participating in regulating local inflammatory responses. MAC exerts a cytolytic effect, directly killing pathogens and causing damage to host cells.
Under physiological conditions, complement activation and regulation are in a balanced state, playing an important role in immune surveillance and regulating self-immune homeostasis; under pathological conditions, an excessively activated complement system attacks self-cells and tissues, leading to various inflammatory responses and autoimmune diseases.
(2) CMKD
CMKD can be divided into two main categories: (1) kidney diseases directly mediated by abnormal activation of the complement system, including C3 glomerulopathy, atypical hemolytic uremic syndrome (aHUS), and immune complex-mediated membranoproliferative glomerulonephritis (IC-MPGN); (2) kidney diseases caused by the involvement of the complement system, including IgA nephropathy, ANCA-associated vasculitis (AAV), diabetic nephropathy, membranous nephropathy, focal segmental glomerulosclerosis (FSGS), lupus nephritis, and antiphospholipid syndrome.
(3) Complement-Related Tests
These include levels of complement and cleavage products in serum, urine, and kidney tissue, complement activity, complement autoantibodies, and complement gene testing (Table 1).

Complement component testing is mostly performed using immunoturbidimetry and enzyme-linked immunosorbent assays; flow cytometry can detect complement regulatory proteins and complement receptors on cell membranes. Measurement of complement activity reflects overall complement function, including complement activation through different pathways to the formation of terminal product MAC. Hemolytic assays (total complement activity CH50 or alternative pathway activity AH50) or enzyme-linked immunosorbent assays are commonly used for detection. Currently, CH50 has been used to monitor the effectiveness of complement blockade therapy (such as eculizumab), with a CH50 value of less than 10% indicating that complement system activation has been effectively blocked. The detection methods for autoantibodies against complement factors or regulatory proteins are primarily immunoturbidimetry and enzyme-linked immunosorbent assays. Immunofluorescence or immunohistochemistry can detect various complement components deposited in kidney tissue, such as C3c, C1q, C4d, MAC, etc.
The collection of complement testing samples must adhere to the following principles: complement testing uses collection tubes containing ethylenediaminetetraacetic acid (EDTA), and complement activity testing uses non-anticoagulant tubes, which should be kept at 4 °C. Plasma/serum should be separated immediately, and if not used immediately, it should be stored at -80 °C within 1 hour to avoid repeated freeze-thaw cycles.
Complement gene testing is primarily based on next-generation sequencing platforms (NGS) for whole-exome sequencing (WES), while copy number variations are detected using multiplex ligation-dependent probe amplification (MLPA).
(4) Complement Targeted Therapy
With the continuous deepening of understanding the role of complement activation in the pathogenesis of CMKD, the complement system has become an important intervention target for this type of disease.
Antigen-antibody complexes, pathogen surface glycoproteins, and abnormal complement regulation can all cause abnormal complement activation, with infection, surgery, and pregnancy being the main triggers. The mechanisms of complement injury mainly include: activation of inflammatory cell activation and tissue damage caused by C3a-C3aR and C5a-C5aR pathways; formation of MAC leading to cell damage.
The targets of complement therapy are related to the role of complement in the pathogenesis of CMKD, including the mannose-binding lectin pathway (MASP-2), the alternative pathway (C3, complement factor B, Bb, complement factor D), and the terminal pathway (C5, C5a, C5aR) and other complement proteins or regulatory proteins. Currently, three drugs have been approved for the treatment of CMKD: eculizumab, ravulizumab, and avacopan. C5 inhibitors eculizumab and ravulizumab have been approved by the U.S. Food and Drug Administration (FDA) for the treatment of aHUS; eculizumab has also been approved by the National Medical Products Administration (NMPA) for the treatment of aHUS; the small molecule C5aR inhibitor avacopan has been approved by the FDA for the treatment of AAV. Several ongoing clinical studies on complement-targeted therapy for CMKD are in progress, including crovalimab, iptacopan, narsoplimab, NM8074, pegcetacoplan, danicopan, ARO-C3, vilobelimab, BDB-001, STSA-1002, IONIS-FB-LRx, cemdisiran, CM338, KP104, ALXN2050, ALXN1720, and HRS-5965.
2. Vaccines
(1) Introduction
Neisseria meningitidis, Streptococcus pneumoniae, and Haemophilus influenzae are the most common pathogens causing invasive infections such as bacterial meningitis and bacteremia. The most significant characteristic of these bacteria is the presence of a capsule, which serves to protect the bacteria from phagocytosis by the body’s innate immune cells while also activating complement; all three activation pathways can form MAC to eliminate bacteria. Therefore, when complement components are deficient or levels are reduced, especially when terminal complement molecules (C5-C9) are absent, the risk of infection by these bacteria increases. Studies have shown that patients with complement deficiencies have a 10,000-fold increased risk of meningococcal infections, and the risk increases 2,000-fold in patients using complement inhibitors.
Although innate immunity is essential, adaptive immune responses are key to preventing these bacterial infections. Epidemiological studies indicate that the susceptible population for Neisseria meningitidis and the periodic outbreaks of epidemic meningitis are primarily due to a lack of antibodies against meningococcus. To this end, the Advisory Committee on Immunization Practices (ACIP) in the United States has specifically proposed vaccination strategies for patients with complement deficiencies or those using complement inhibitors, recommending that patients using complement inhibitors receive the B-type meningococcal vaccine and the ACYW group meningococcal polysaccharide vaccine at least two weeks before starting the drug; it is recommended that patients with complement deficiencies, especially those with C1, C2, C3, and C4 deficiencies, receive the pneumococcal vaccine.
(2) Vaccination Recommendations
This consensus recommends vaccination with meningococcal and pneumococcal vaccines for users of complement inhibitors based on the availability of vaccines in China, the epidemiological status of Neisseria meningitidis, Streptococcus pneumoniae, and Haemophilus influenzae serotypes, and related guidelines from abroad (specific requirements for vaccination with different targeted complement inhibitors can be referred to in the drug instructions):
1. Meningococcal vaccine:
(1) Vaccination time: It is recommended to vaccinate at least two weeks before treatment with complement inhibitors;
(2) Vaccine type: The ACYW135 group meningococcal polysaccharide vaccine (MPV-ACYW) or the ACYW135 group meningococcal polysaccharide conjugate vaccine (MPCV-ACYW) can be chosen;
(3) Number of doses: 1 dose;
(4) Revaccination: Depending on the treatment needs of complement inhibitors, polysaccharide vaccines can be revaccinated once after three years, and conjugate vaccines can be revaccinated once after five years.
2. Pneumococcal vaccine:
(1) Starting vaccination time: It is recommended to vaccinate at least two weeks before treatment with complement inhibitors;
(2) Vaccine type: The 13-valent pneumococcal conjugate vaccine (PCV13) and the 23-valent pneumococcal polysaccharide vaccine (PPV23) can be chosen;
(3) Number of doses: 2 doses;
(4) Immunization procedure: The first dose should be PCV13, followed by a second dose of PPV23 after 12 months.
(3) Remarks
1. Immunization planning for eligible children should follow the provisions of the “National Immunization Program Vaccine Children Immunization Procedures and Instructions (2021 Edition)” and the “Guiding Principles for the Use of Non-Immunization Program Vaccines (2020 Edition)”. Specific immunization methods can refer to the “Chinese Expert Consensus on Meningococcal Vaccine Prevention (2023 Edition)”.
2. In emergency situations, patients who have received the meningococcal vaccine and start treatment with complement inhibitors less than two weeks later must receive appropriate prophylactic sensitive antibiotic treatment until the vaccination period reaches two weeks.
3. CMKD
(1) aHUS
aHUS is a type of complement-mediated thrombotic microangiopathy (TMA). TMA is a group of clinical-pathological syndromes characterized by microangiopathic hemolytic anemia (MAHA), thrombocytopenia, and multiple organ damage. In addition to aHUS, primary TMA also includes Shiga toxin-producing Escherichia coli-associated hemolytic uremic syndrome (STEC-HUS) and thrombotic thrombocytopenic purpura (TTP). The incidence of aHUS is 0.23 to 1.9 cases per million, with infection and pregnancy being the main triggers. Patients with aHUS have a poor prognosis; in the pre-complement treatment era, the mortality rate during the acute phase was 25%, and about 50% of patients progressed to end-stage kidney disease within a year.
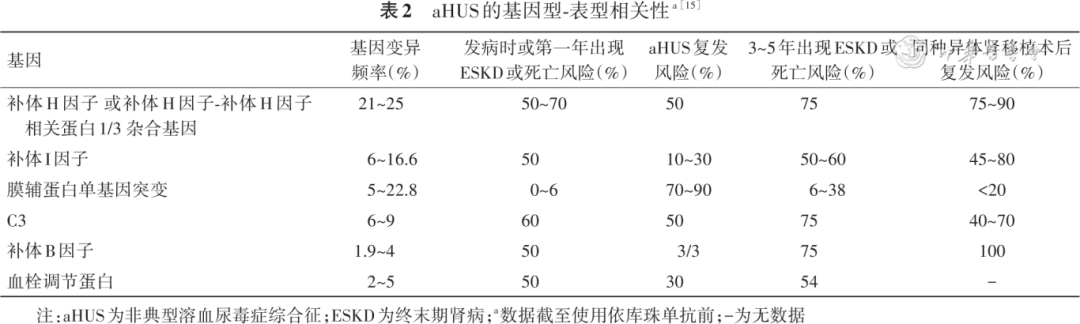
About 50% of aHUS patients have potential genetic and/or acquired abnormalities in the complement system, with the pathogenesis mainly involving mutations in complement regulatory protein genes, or mutations in complement inherent protein genes (Table 2), or antibodies against complement regulatory proteins, triggered by events leading to excessive activation of the alternative pathway, resulting in MAC formation, causing endothelial injury, coagulation cascade activation, and clinical manifestations of multi-organ damage in the kidneys, nervous system, cardiovascular system, gastrointestinal tract, and respiratory system.
aHUS is a diagnosis of exclusion. First, TMA is diagnosed based on the patient’s thrombocytopenia (platelet count <150×109/L or a decrease of >25% from baseline), microangiopathic hemolytic anemia (peripheral blood smear shows >1% fragmented red blood cells, lactate dehydrogenase elevated >1.5 times the upper limit of normal reference value, combined with globulin reduction, hemoglobin reduction), and organ damage (kidneys, nervous system, cardiovascular system, gastrointestinal tract, respiratory system, etc.). Further exclude TTP and STEC-HUS, and finally diagnose aHUS (Figure 1). All cases of aHUS caused by secondary factors should receive supportive treatment, and after completing relevant examinations, treat the primary disease. If TMA continues to progress after controlling the primary disease, consider the possibility of complement-mediated aHUS. After considering the diagnosis of aHUS, further complement-related gene, complement level, function, and anti-complement antibody testing (see “Complement-Related Tests”) can be conducted for prognostic assessment and to guide long-term treatment plans.
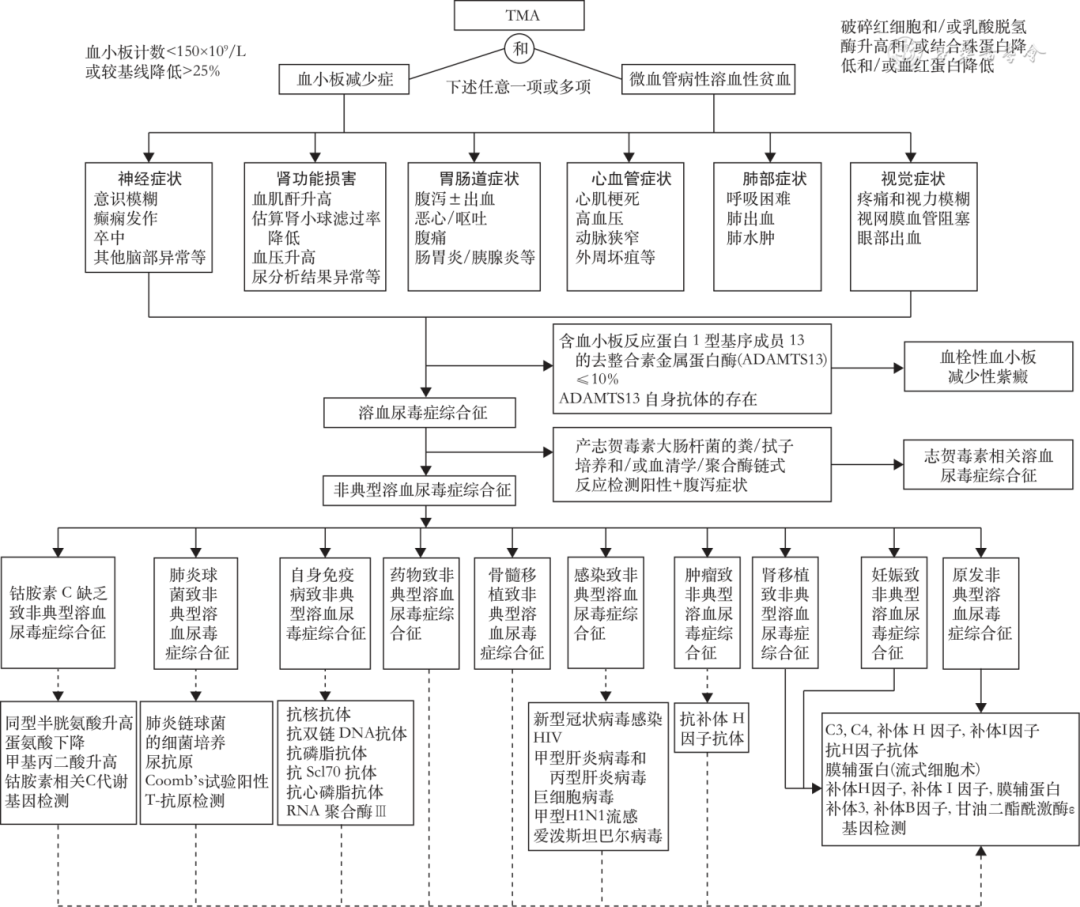
▲Figure 1 Diagnosis flowchart for thrombotic microangiopathy (TMA) mediated by atypical hemolytic uremic syndrome
Patients suspected of aHUS can initiate plasma exchange, but due to the lack of evidence for improving long-term kidney prognosis, it is recommended that patients diagnosed with aHUS use complement inhibitors as early as possible, and those with positive complement factor H antibodies should receive immunosuppressive treatment, with antibody titers monitored as indicators. Eculizumab has been approved for aHUS indications in China. Ravulizumab was approved by the FDA for the treatment of aHUS in 2019. Other global multicenter clinical studies on complement-targeted therapy for aHUS, such as iptacopan (NCT04889430), NM8074 (NCT05684159), and crovalimab (NCT04861259), are still ongoing.
The treatment goal for aHUS is to achieve TMA remission as quickly as possible, without early relapse. After diagnosing complement-mediated aHUS, it is essential to initiate treatment with eculizumab within 24 to 48 hours of onset to control disease progression (Figure 2). Phase II clinical studies of eculizumab for aHUS have shown that rapid suppression of complement activity occurs within one hour of the first dose of eculizumab. After 7 days of treatment, platelet counts significantly increased; after one month of treatment, 57.1% of patients improved serum creatinine, and after a median of 5 treatments, patients achieved complete complement suppression, with a complete kidney remission rate of 47.8%, better than those who did not use it (14.3%); after 26 weeks of treatment, 83% (20/24) of patients were off dialysis; within two years, 76% of patients improved kidney function, and 76% achieved complete TMA remission.
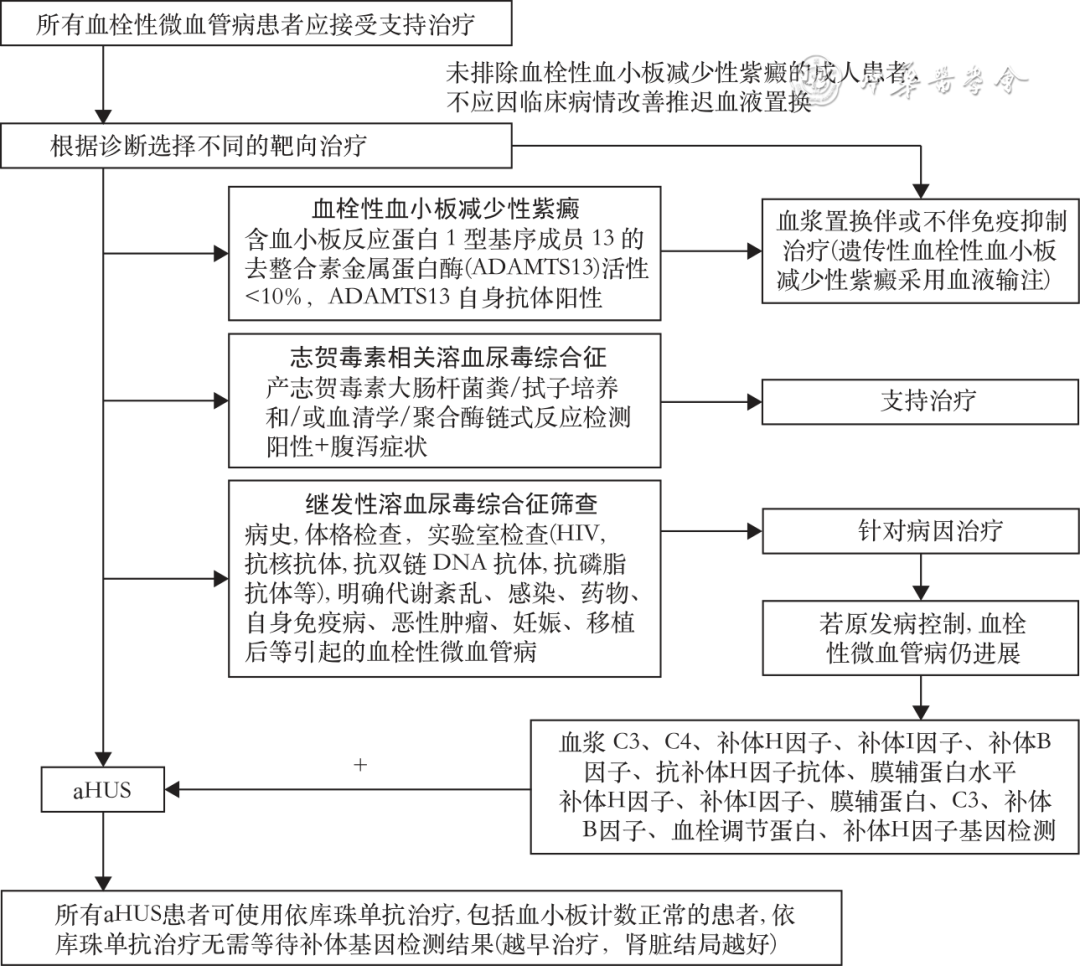
▲Figure 2 Diagnosis and treatment flow for adult atypical hemolytic uremic syndrome (aHUS) (acan be detected by Sanger method or next-generation sequencing technology)
The expert consensus developed by the Kidney Disease: Improving Global Outcomes (KDIGO) on the treatment of atypical hemolytic uremic syndrome and C3 glomerulopathy indicates that all complement-related aHUS patients should be treated for at least 6 to 12 months, and consideration can be given to discontinuing complement inhibitors after kidney function has returned to baseline (or stabilized in cases of chronic kidney disease) for at least three months, depending on individual patient circumstances (genetic mutations, transplant history, relapse history, dialysis history, etc.). The duration of treatment remains controversial.
(2) C3 Glomerulopathy
C3 glomerulopathy is a rare kidney disease mediated by excessive activation of complement, including dense deposit disease (DDD) and C3 glomerulonephritis. The incidence of C3 glomerulopathy globally is 1 to 2 cases per million per year. About 50% of patients progress to end-stage kidney disease within 10 years, and 50% to 70% of patients experience recurrence after kidney transplantation.
C3 glomerulopathy is associated with dysregulation of the complement alternative pathway, with about 25% of patients having mutations in complement genes, such as those encoding C3, complement factor H, complement factor I, MCP, and CFHR5. C3 nephritis factors (C3NeF) are present in serum in 80% of DDD and more than 50% of C3 glomerulonephritis patients. Diagnosis of C3 glomerulopathy is made through renal tissue biopsy pathology, with diagnostic criteria being an intensity of C3c immunofluorescence greater than that of other immunoglobulin fluorescence ≥2+, while excluding post-infectious glomerulonephritis and other diseases, with confirmation requiring integration of immunopathology, light microscopy, electron microscopy, and clinical presentation. C3 glomerulopathy shows diverse appearances under light microscopy, and electron microscopy is used for classification of DDD and C3 glomerulonephritis. Given the possibility of immunoglobulin deposition being obscured, leading to misdiagnosis of other kidney diseases as C3 glomerulonephritis, such as proliferative glomerulonephritis with monoclonal immunoglobulin deposition (PGNMID), it is recommended that all patients with suspected C3 glomerulonephritis (especially adults) undergo IgG and light chain staining on paraffin sections digested with streptokinase. For patients clinically suspected of C3 glomerulopathy, complement-related testing should be performed, such as C3, C3 nephritis factors, autoantibodies against complement factor B, and autoantibodies against complement factor H (see “Complement-Related Tests”).
C3 glomerulopathy must also be differentiated from other types of glomerulonephritis, including post-infectious glomerulonephritis (PIGN), immune complex-mediated membranoproliferative glomerulonephritis, and monoclonal gammopathy of renal significance (MGRS) (Table 3).

The treatment principles for C3 glomerulopathy include blood pressure control, with priority given to angiotensin-converting enzyme inhibitors (ACEi) or angiotensin receptor blockers (ARB); for patients with urinary protein still greater than 500 mg/24h after supportive treatment or with recent elevations in serum creatinine indicating a risk of disease progression, or if renal biopsy pathology shows moderate to severe inflammation, it is recommended to use glucocorticoids combined with mycophenolate mofetil as the initial treatment; if the efficacy is poor, consider using eculizumab; if treatment fails, consider combination therapy with other complement-targeted drugs. Phase II clinical studies of iptacopan for C3 glomerulopathy have shown good efficacy and safety, with an average estimated glomerular filtration rate (eGFR) improvement of 1.04 ml·min-1·1.73 m-2 at week 12, and extending treatment to 12 months can further reduce urinary protein (decreased by 57% from baseline) and improve eGFR (6.83 ml·min-1·1.73m-2). For patients with recurrent C3 glomerulopathy after transplantation, long-term treatment with iptacopan can stabilize eGFR. Phase II clinical studies of avacopan for C3 glomerulopathy have shown that treatment over 26 weeks can improve chronic pathology scores of C3 glomerulopathy and significantly improve eGFR. Phase II clinical studies of pegcetacoplan for C3 glomerulopathy have shown that urinary protein can decrease by more than 65% after 48 weeks of treatment.
(3) Immune Complex-Mediated Membranoproliferative Glomerulonephritis (IC-MPGN)
IC-MPGN is a rare pathological type characterized by the formation of double track signs in the capillary walls of renal tissue under light microscopy. IC-MPGN can be divided into monoclonal immunoglobulin-associated MPGN (membranoproliferative glomerulonephritis) and polyclonal immunoglobulin-associated MPGN (including autoimmune diseases, infections, etc.).
The characteristic of IC-MPGN is the formation of antigen-antibody immune complexes leading to activation of the classical pathway of complement, resulting in deposition of immune complexes and complement activation fragments in the glomeruli. Diagnosis of idiopathic IC-MPGN requires evaluation of potential diseases in IC-MPGN patients, excluding infections, autoimmune diseases, monoclonal gammopathy, and fibrous glomerulonephritis. About 50% of idiopathic IC-MPGN patients have abnormal activation of the complement alternative pathway. Some patients have mutations in complement genes, and 40% to 50% of IC-MPGN patients have complement nephritis factors, C5NeF, anti-complement factor H antibodies, anti-complement factor B antibodies, or anti-C3b antibodies.
After clarifying the etiology of IC-MPGN, initial treatment should target the underlying cause. Whether idiopathic-related IC-MPGN or associated with primary disease, the best treatment method is supportive therapy, with cautious use of immunosuppressants. For idiopathic IC-MPGN with nephrotic syndrome, a single-arm clinical study of eculizumab in treating MPGN (EAGLE study) suggests that some patients may achieve partial remission with eculizumab treatment.
(4) AAV
AAV is a group of systemic diseases characterized by necrotizing vasculitis. In China, myeloperoxidase anti-neutrophil cytoplasmic antibody-associated glomerulonephritis (MPO-ANCA) is predominant, with the kidneys being one of the most commonly affected organs.
In patients with AAV, the concentrations of C3a, C5a, and Bb in circulation and urine during the acute phase are significantly higher than during remission, and the concentration of Bb is correlated with disease activity, suggesting that the activation of the complement alternative pathway is involved in the pathogenesis of AAV. Animal experiments have shown that C5a and its receptors are particularly critical in stimulating neutrophil activation and vascular injury.
The small molecule C5aR antagonist avacopan has been approved by the FDA for the treatment of AAV. Phase II clinical studies of avacopan for AAV (CLEAR study) show that the remission rate of avacopan treatment at week 12 is not inferior to that of the prednisone group. Phase III clinical studies of avacopan for AAV (ADVOCATE study) show that the remission effect of avacopan at week 26 is not inferior to that of the prednisone reduction scheme, and the sustained remission effect at week 52 is better than that of the prednisone reduction scheme, with significant improvement in eGFR. Ongoing clinical studies targeting C5a, such as vilobelimab for AAV, have completed Phase II studies, while STSA-1002 and BDB-001 are undergoing Phase I/II studies for treating AAV.
(5) IgA Nephropathy
IgA nephropathy is the most common primary glomerulonephritis in China. Approximately 25 new cases of IgA nephropathy are diagnosed annually per million adults worldwide, particularly prevalent among East Asian populations. The deposition of IgA molecules in the mesangial region of the glomeruli is a characteristic pathological manifestation of IgA nephropathy patients. About 20% to 50% of IgA nephropathy patients progress to end-stage kidney disease within 10 to 20 years.
The pathogenesis of IgA nephropathy is currently believed to involve the “four-hit” hypothesis: the first hit is the elevation of galactose-deficient IgA1 (Gd-IgA1), the second hit is the production of specific antibodies against Gd-IgA1 in circulation, the third hit is the formation of pathogenic immune complexes containing Gd-IgA1; the fourth hit is the deposition of pathogenic immune complexes in the mesangial region of the glomeruli, activating the complement system, inducing inflammatory responses, and causing glomerular injury.
The complement system plays an important role in the pathogenesis of IgA nephropathy. In IgA nephropathy, complement activation is primarily through the alternative pathway, followed by the lectin pathway. Various complement protein levels and genetic variations are associated with the occurrence, severity, and/or progression of IgA nephropathy, including increased deposition of glomerular C3, C4d, MBL, CFHR5, etc.; elevated ratios of IgA/C3, Gd-IgA1/C3, CFHR5, CFHR1 in circulation, and decreased levels of C3, MASP-3, etc.; increased levels of C3a, C5a in urine, and decreased levels of complement factor H; genetic variations affecting gene expression or protein function of complement factor H, CFHR5, MBL2, C3, and other genes; and genetic deletions of CFHR3-CFHR1.
Currently, several complement-targeted drugs are undergoing Phase II/III clinical studies, including the C5aR antagonist avacopan, complement factor B inhibitors iptacopan, and MASP-2-targeting narsoplimab. A randomized, double-blind, placebo-controlled Phase II clinical study of iptacopan for treating IgA nephropathy showed that after three months (46 cases) or six months (66 cases) of treatment, the urinary protein-creatinine ratio (UPCR) decreased by 31% and 41%, respectively. The Phase II clinical study of narsoplimab for treating IgA nephropathy also demonstrated its effect in reducing urinary protein, but its Phase III clinical study (ARTEMIS-IgAN study) did not reach the primary endpoint of reducing urinary protein and was prematurely terminated.
(6) Lupus Nephritis
Lupus nephritis is the most common complication of systemic lupus erythematosus and is also the most common secondary glomerulonephritis in China. The complement system plays a “double-edged sword” role in the pathogenesis of lupus nephritis; early genetic variations in complement components can increase disease susceptibility; while lupus nephritis involves the activation of three complement pathways, excessive activation causes organ damage.
Anti-C1q A08 autoantibodies are associated with disease activity and prognosis in lupus nephritis, and in vitro experiments have shown that anti-C1q A08 antibodies play an important role in activating the classical pathway of complement; studies have indicated that MBL-2 gene polymorphisms are related to MBL plasma concentrations and are also associated with multi-system involvement in systemic lupus erythematosus; defects in complement alternative pathway components, such as genetic variations in complement factor H and its related proteins, increase susceptibility to systemic lupus erythematosus. Compared to patients with systemic lupus erythematosus without kidney involvement, lupus nephritis patients have higher plasma Bb levels, suggesting that alternative pathway activation plays a more important role in the pathogenesis of lupus nephritis.
About 20% of lupus nephritis patients have kidney TMA. Previous studies have shown that C4d deposition in glomeruli and decreased serum complement factor H in lupus nephritis patients with TMA are associated with poor kidney outcomes. Lupus nephritis patients with TMA have significantly higher plasma Ba and urinary Ba levels than those without TMA, indicating more pronounced alternative pathway activation in lupus nephritis with TMA.
The KDIGO guidelines for lupus nephritis recommend that patients with lupus nephritis and TMA should be treated based on the potential causes of TMA, and the activity and antibodies of the disintegrin and metalloproteinase with a thrombospondin type 1 motif member 13 (ADAMTS13) should be tested. For primary or secondary complement-mediated TMA with normal ADAMTS13 activity and negative antiphospholipid antibodies, eculizumab treatment may be considered. Currently, complement factor B inhibitors iptacopan, C5-targeted drugs ravulizumab, and MASP-2-targeted drugs narsoplimab are undergoing Phase II clinical trials and may become new treatment options for lupus nephritis.
(7) Antiphospholipid Syndrome
Antiphospholipid syndrome is an autoimmune disease characterized by recurrent venous or arterial thrombosis and pathological pregnancy, with persistent moderate to high positivity of antiphospholipid antibodies in serum. Antiphospholipid antibodies mediate the occurrence and development of antiphospholipid syndrome through various pathways, with excessive complement activation participating in its pathogenesis. Antiphospholipid antibodies can induce activation of the classical pathway of complement, activating endothelial cells, leading to increased vascular permeability, selective expression of adhesion molecules and chemokines, and subsequently causing thrombosis; complement activation products C5a can stimulate neutrophils and monocytes to produce tumor necrosis factor, transcription factors, and soluble vascular endothelial growth factor receptor 1, all of which can impair placental function. Additionally, complement can also activate the coagulation system to induce thrombosis. Patients with catastrophic antiphospholipid syndrome have complement gene variations, and both the classical and alternative pathways of complement are significantly activated.
Treatment for antiphospholipid syndrome should be individualized, with anticoagulation being the standard treatment for thrombotic antiphospholipid syndrome; the combination of aspirin and low molecular weight heparin is the main treatment method for obstetric antiphospholipid syndrome. Complement blockade may be an effective treatment for severe antiphospholipid antibody-related complications during pregnancy. A retrospective study of eculizumab for treating catastrophic antiphospholipid syndrome analyzed the efficacy and safety of eculizumab in 39 cases (6.7%), of which 30 cases were for salvage therapy and 9 cases for first-line treatment, showing that 29 cases (74.4%) had significant improvement.
(8) Membranous Nephropathy
Membranous nephropathy is a group of diseases characterized by the deposition of immune complexes beneath the epithelial cells of the glomerular basement membrane, accompanied by thickening of the basement membrane. About 70% of patients with primary membranous nephropathy are positive for serum anti-M-type phospholipase A2 receptor (PLA2R) antibodies. In 1980, Salant et al. discovered that complement activation is one of the necessary conditions for kidney injury in the Heymann nephritis model. Most anti-PLA2R antibodies are of the IgG4 subtype. In 2021, Haddad et al. found that anti-PLA2R-IgG4 directly binds to MBL in a glycosylation-dependent manner, activating the complement system; specific blockade of MBL can reverse this effect. Additionally, the alternative pathway is also involved in the complement activation mechanism of primary membranous nephropathy.
The treatment of membranous nephropathy mainly includes supportive treatment and immunosuppressive therapy. Phase I/II clinical studies targeting C3, C5, and MASP are ongoing. However, clinical studies of eculizumab for treating membranous nephropathy have not shown significant efficacy. Clinical studies targeting complement factor B, complement factor D, and other alternative pathway complement proteins have also been prematurely terminated due to lack of significant efficacy. Recent studies have found that administering C3aR antagonists in a rat model of membranous nephropathy significantly reduces urinary protein and kidney tissue pathological damage; inhibiting the binding of C3a to C3aR on podocyte surfaces can alleviate podocyte apoptosis and dysfunction; reduce the production of disease-associated specific IgG and its deposition in kidney tissue and subsequent complement activation, without affecting overall IgG levels.
(9) FSGS
FSGS is one of the main types of nephrotic syndrome, characterized by sclerosis affecting some glomeruli and capillary loops visible under light microscopy, with podocyte foot process fusion and cell shedding as the main features under electron microscopy.
Research shows that abnormal activation of the classical and alternative pathways of complement may participate in its pathogenesis, with complement activation products detectable in urine, while abnormalities in complement regulatory proteins may be involved in podocyte injury and the progression of FSGS. In patients with primary FSGS, lower plasma C3 or higher urinary C3a and C5a levels are associated with higher urinary protein levels, higher serum creatinine, and poorer kidney prognosis. Urinary Bb concentration is an independent risk factor for ineffective treatment and end-stage kidney disease. Patients with glomeruli showing simultaneous deposition of IgM and C3 in renal tissue fluorescence have poorer treatment response and kidney prognosis. The percentage of C4d-positive glomeruli in FSGS patients is significantly higher than in minimal change disease. In FSGS rat models, urinary protein is present without progressing to FSGS lesions, with an increased percentage of C4d-positive glomeruli, suggesting that C4d may play a pathogenic role in FSGS. In mouse models of FSGS, C3-deficient mice show less glomerulosclerosis, milder tubular interstitial injury, and better kidney function; CD59-deficient mice exhibit more severe glomerulosclerosis and tubular interstitial injury, suggesting that complement activation is involved in the occurrence and development of kidney injury in FSGS. Currently, drugs inhibiting complement activation are in active development, with C5-targeted FSGS treatment undergoing Phase I clinical trials.
(10) Diabetic Nephropathy
Diabetic nephropathy is one of the most common and serious complications of diabetes, becoming the leading cause of chronic kidney disease patients hospitalized in China. Rasmussen et al. found that elevated baseline plasma C3 is associated with an increased risk of diabetic nephropathy in healthy individuals, suggesting that complement activation participates in the occurrence and development of the disease. Clinical studies have found that multiple complement proteins such as C3a, C5a, and Bb are upregulated in circulation and urine in patients with diabetic nephropathy, closely related to disease severity, and patients with C3c and C1q deposition in kidney tissue have worse kidney outcomes.
Animal experiments have shown that C3aR-/- mice have less severe diabetic kidney injury, with significant suppression of inflammatory responses and T cell adaptive immunity; C5a/C5aR1 signaling mediates the occurrence and progression of kidney diseases by altering mitochondrial metabolism, while the absence of complement factor B can alleviate tubular cell injury.
There are two mechanisms of abnormal complement activation in diabetic nephropathy: (1) hyperglycemia stimulates the formation of glycosylated proteins on cell membranes, activating the mannose-binding lectin pathway; (2) hyperglycemia leads to abnormal glycosylation of complement membrane regulatory protein CD59, insufficiently inhibiting MAC formation, resulting in the cytotoxic effects of complement. The pathogenic mechanism of abnormal complement activation may be regulating inflammation and lipid metabolism.
C5-targeted drug ALXN1720 is undergoing Phase I clinical research for treating diabetic nephropathy, and drugs targeting complement factor B, C3a, C5a, and their receptors may become future therapeutic agents for diabetic nephropathy.
4. Recommendations
Despite significant progress in understanding the mechanisms of complement involvement in CMKD, current diagnosis and treatment status, and complement-targeted therapies, many aspects still require further research and improvement.
(1) Complement-Related Testing
Currently, only a few foreign laboratories can perform comprehensive complement testing. International quality control results of complement testing show that only a few laboratories can standardly measure complement components, such as C3, C4, and anti-complement factor H antibody concentrations, with only half of the laboratories successfully detecting complement activation levels in test samples.
China still lacks relevant testing institutions; for example, C3 nephritis factors are only detected through enzyme-linked immunosorbent assays, with low positive rates. In contrast, foreign reports indicate that combining 4 to 5 methodologies can achieve positive rates of 50% to 80%. Therefore, improving various methodologies is key to ensuring testing sensitivity.
Testing for complement components, complement antibodies, complement function, and complement genes needs to establish corresponding testing platforms in China to support disease diagnosis and efficacy monitoring.
Due to the complex sequence structures of important complement protein genes such as CFHRs, C4, and complement receptor 1, next-generation sequencing technology cannot detect these genes, thus the development and application of third-generation sequencing is also a focus of future complement gene testing.
(2) Complement-Related Biomarkers
For different CMKDs, whether levels of complement inherent proteins, regulatory proteins, complement activation products, and complement-related autoantibodies can serve as corresponding biological markers in diagnosis, treatment, and efficacy monitoring still requires further research. Currently, only a few complement-related indicators have clear clinical application value, such as CH50, complement factor H antibodies, C3 nephritis factors, etc.
(3) Mechanisms of Complement Involvement in Pathogenesis
The activation of complement in various CMKDs involves different pathways, different molecules, and varying degrees of activation. Various complement molecules participating in different pathways can serve as targets for targeted therapy. Therefore, clarifying the complement activation pathways and key activated molecules in different CMKDs is crucial for selecting complement-targeted drugs in the future, necessitating more clinical, in vivo (animal experiments), in vitro (cell experiments), and genomics research.
(4) Safety of Complement-Related Drugs
In recent years, complement-targeted drugs have gradually been applied in clinical practice, and clinicians have limited knowledge of these drugs, necessitating further observation of their safety. Prior to anti-C5 monoclonal antibody treatment, relevant vaccines must be administered, primarily concerned that blocking common pathways of complement activation may lead to infections by encapsulated bacteria such as Neisseria meningitidis, Streptococcus pneumoniae, and Haemophilus influenzae. However, more and more complement-targeted drugs can accurately target specific molecules in specific complement pathways, and whether relevant vaccine injections should also be performed before use requires further research.
Consensus Expert Group List
Expert Group Leader: Zhao Minghui (Department of Nephrology, Peking University First Hospital)
Expert Group Members (sorted by surname in pinyin): Bu Fengxiao (Data Science Department, Sichuan University West China Hospital Rare Disease Research Institute); Chen Min (Department of Nephrology, Peking University First Hospital); Cui Zhao (Department of Nephrology, Peking University First Hospital); Fu Ping (Department of Nephrology, Sichuan University West China Hospital); Hao Chuanming (Department of Nephrology, Fudan University Huashan Hospital); Li Ke (Laboratory, Second Affiliated Hospital of Xi’an Jiaotong University); Li Wenge (Department of Nephrology, China-Japan Friendship Hospital); Li Xuemei (Department of Nephrology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences); Liu Zhangsuo (Nephrology Research Center, First Affiliated Hospital of Zhengzhou University); Lv Jicheng (Department of Nephrology, Peking University First Hospital); Mao Yonghui (Department of Nephrology, Beijing Hospital); Tan Ying (Department of Nephrology, Peking University First Hospital); Wang Weiming (Department of Nephrology, Ruijin Hospital Affiliated to Shanghai Jiao Tong University School of Medicine); Wang Yue (Department of Nephrology, Peking University Third Hospital); Wu Jiang (Chinese Center for Disease Control and Prevention); Yu Feng (Department of Nephrology, Peking University International Hospital); Yu Xiaojian (Department of Nephrology, Peking University First Hospital); Zhang Hong (Department of Nephrology, Peking University First Hospital); Zhou Fude (Department of Nephrology, Peking University First Hospital); Zhu Li (Department of Nephrology, Peking University First Hospital); Zuo Li (Department of Nephrology, Peking University People’s Hospital)
This article is sourced from: Chinese Journal of Internal Medicine, 2024, 63(3): 258-271.
This article is edited by: Hu Zhaohui